Vol 1 | Issue 1 | May – August 2015 | page:23-28 | Yogesh Panchwagh[1*].
Author: Yogesh Panchwagh[1*].
[1]Orthopaedic Oncology Clinic, Pune, India.
Address of Correspondence
Dr. Yogesh Panchwagh.
Orthopaedic Oncology Clinic, 101, Vasant plot 29, Bharat Kunj Society -2, Erandwana, Pune – 38, India.
Email: drpanchwagh@gmail.com
Abstract
Ewing sarcoma is one of the common primary bone sarcomas affecting patients mostly in the second decade. Appropriate clinical examination, investigations, staging, biopsy and multi-modal treatment are essential for good outcome. Neo-adjuvant and adjuvant chemotherapy have shown definite benefits in local and systemic control and in improving survival. Though historically, emphasis of treatment was on radiation, non metastatic Ewing sarcoma is shown to have better outcome with surgical excision as compared to definitive radiotherapy. Limb salvage surgery is currently the norm given the excellent functional outcomes. Various reconstruction options are available depending upon the age, site and size of the lesion. Appropriate follow up is essential to pick up local and systemic failures early. Individualized approach may be required for patients who are metastatic at presentation.
Keywords: Ewing sarcoma, Surgery, Limb salvage, reconstruction.
Introduction
Ewing Sarcoma (ES) is a highly aggressive malignant tumor affecting mostly the immature skeleton, more commonly in the second decade of life. ES is named after Dr. James Ewing, a pathologist. Its aetiopathogenesis has evolved from “Endothelioma of bone” to a unique malignant tumor of bone with well described translocation t(11;22)(q24;q12) as a possible causative factor [1]. The classical pathology of small round blue cells makes it a part of the Round cell family of tumors, the other members of which are rhabdomyosarcoma, synovial sarcoma, non-Hodgkin’s lymphoma, retinoblastoma, neuroblastoma, hepatoblastoma, and nephroblastoma or Wilms’ tumor [2].
Clinical presentation
A high index of suspicion is required to diagnose a primary bony sarcoma like ES at a very early stage. The patients, typically in their first two decades of life, usually have a history of 2-6 months duration, of a painful, progressively increasing swelling in the affected area. Most of the patients give a concomitant history of trauma, which is coincidental. Some of the patients may have a history of fever [3,4].
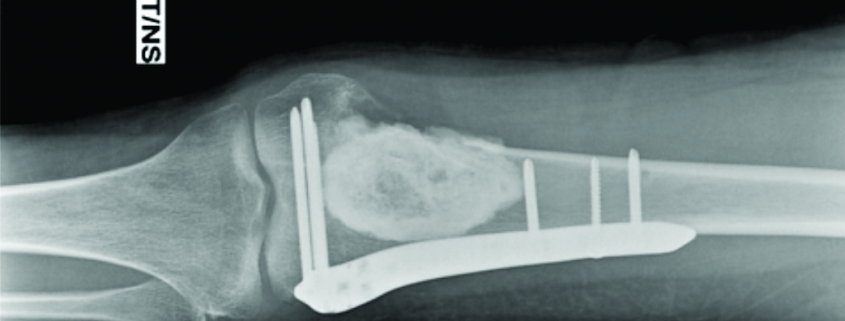
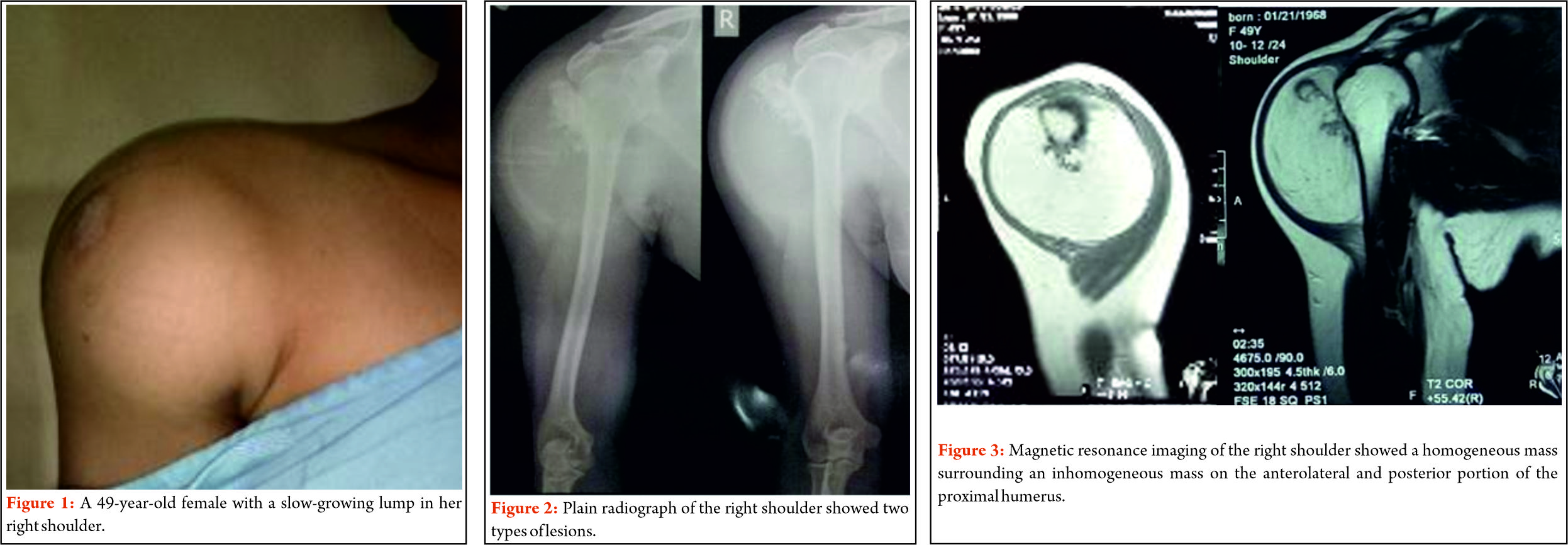
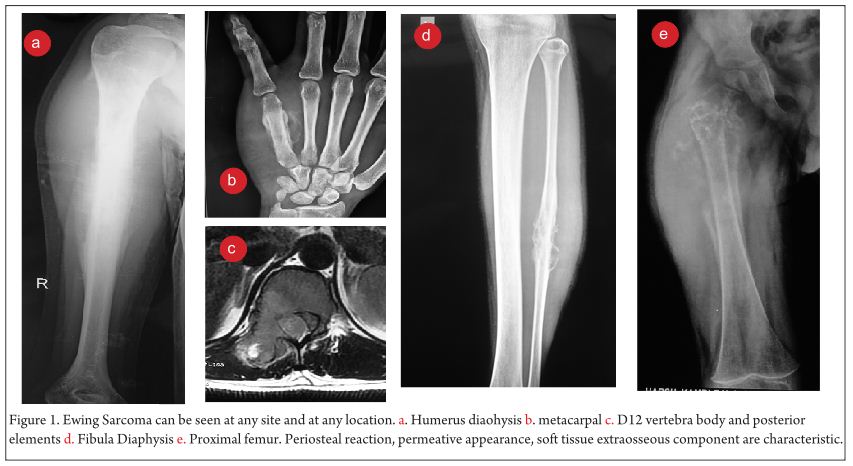
Clinical examination reveals a tender diffuse swelling in the affected area. The range of motion of the adjoining joint may be terminally restricted. The palpation may reveal local warmth. Though the most commonly affected site is the diaphysis in the bone, ES is known to affect the metaphyseal region as well [3]. ES may affect any bone in the body (Fig 1 a-e). Periosteal ES located on the surface of bone and soft tissue ES, though rare, are well-defined clinical entities [3].
‘The clinical and radiological features in an ES of bone may be akin to osteomyelitis or an eosinophilic granuloma and these differentials have to be borne in mind and ruled out by subsequent investigations.
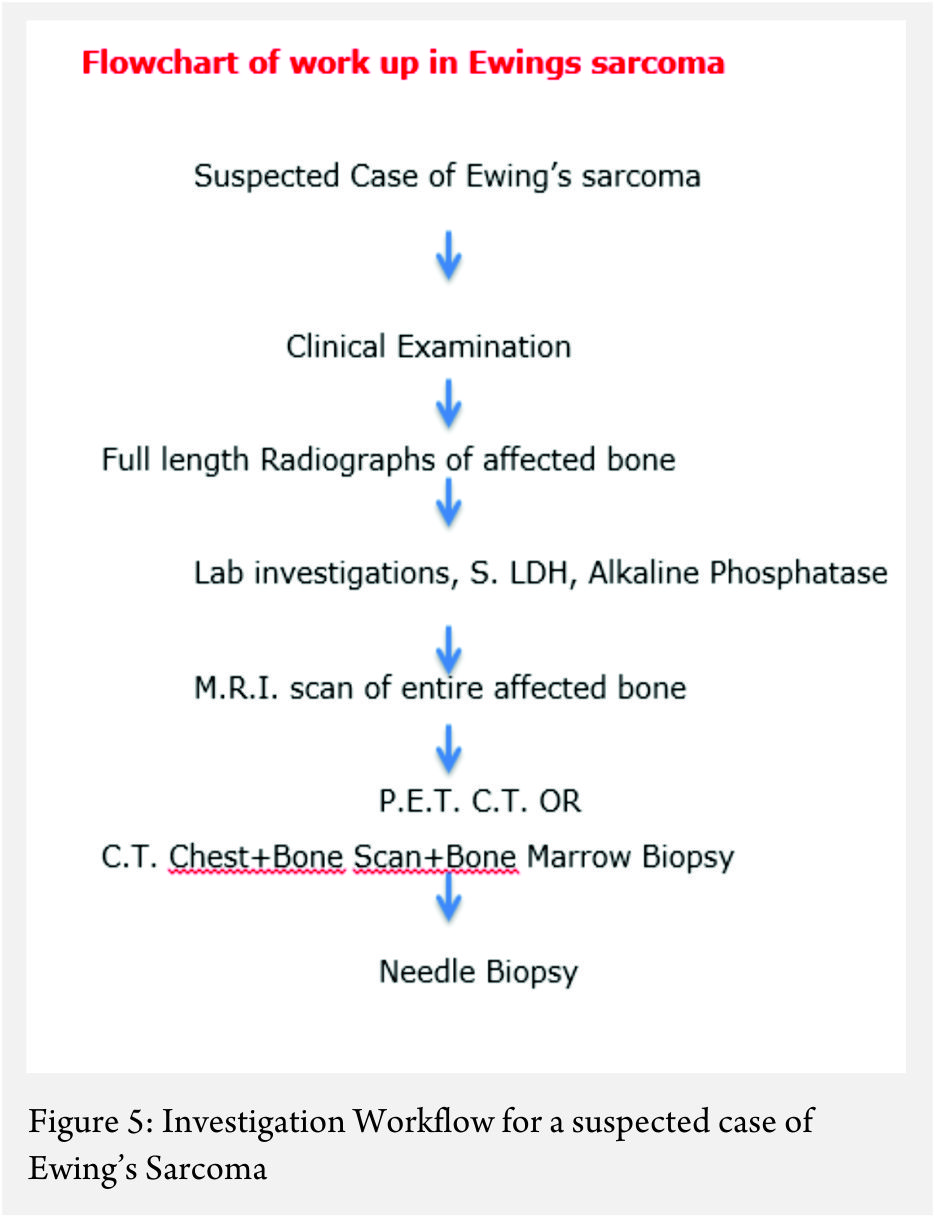
Work up:
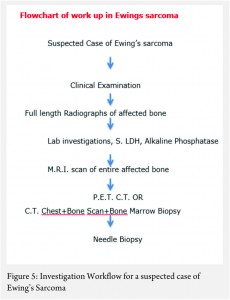
The work up includes plain radiographs of the affected bone including the nearby joint, M.R.I. scan of the involved bone, and either a PET CT [7,8,9] or a CT Chest with Technetium Bone scan and a bone marrow aspiration biopsy [3]. The x ray ( Figure 1, a-e) usually shows a permeative, lytic lesion with lamellated periosteal reaction. In locally advance cases, an extra osseous soft tissue component is common [1,3,5]. A diaphyseal lesion may exhibit a characteristic “Onion peel” periosteal reaction. In some cases, “hair-on-end” or “sun-ray spicule” type of periosteal reaction may also be seen.
The laboratory investigations may reveal leukocytosis with elevated E.S.R. and C.R.P [3]. Serum levels of Lactate Dehydrogenase (S. LDH) are usually elevated and serve as a marker of disease activity and response to treatment [6].
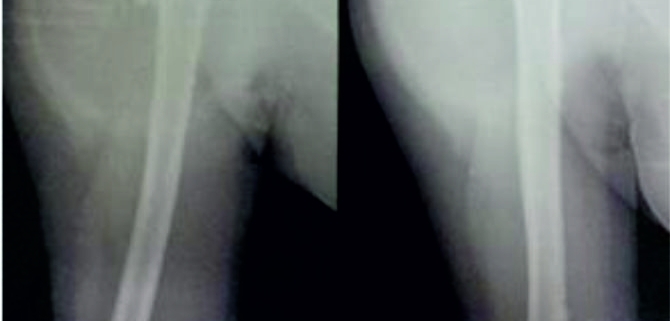
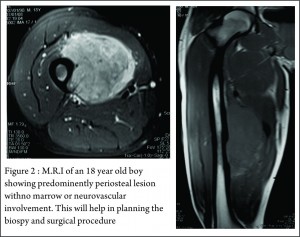
These clinico-radiological and laboratory parameters are akin to osteomyelitis and it requires a trained eye with high index of suspicion to pick the neoplastic nature early in order to avoid mistreating these patients. MRI scan (Fig 2 ) has emerged as one of the most important radiological investigation amongst the others, in the work-up of primary bone sarcomas. It helps immensely in delineating the marrow involvement, revealing skip lesions if any, understanding the extent of soft tissue component and its relationship with the neuro vascular bundle, joint involvement and to decide the ideal site for biopsy. M.R.I. can also be used to assess response to neo-adjuvant chemotherapy [3,5].
Staging
Staging in a case of ES is of paramount importance because of its bearing on the overall prognosis and treatment decisions [1,3]. The conventional staging investigations included a C.T. scan of the chest, a three phase technetium mendronate bone scan and a bone marrow aspiration biopsy [3,10]. However, with the advent of P.E.T. C.T., the bone marrow aspiration biopsy is being found unnecessary [8,9].
Biopsy
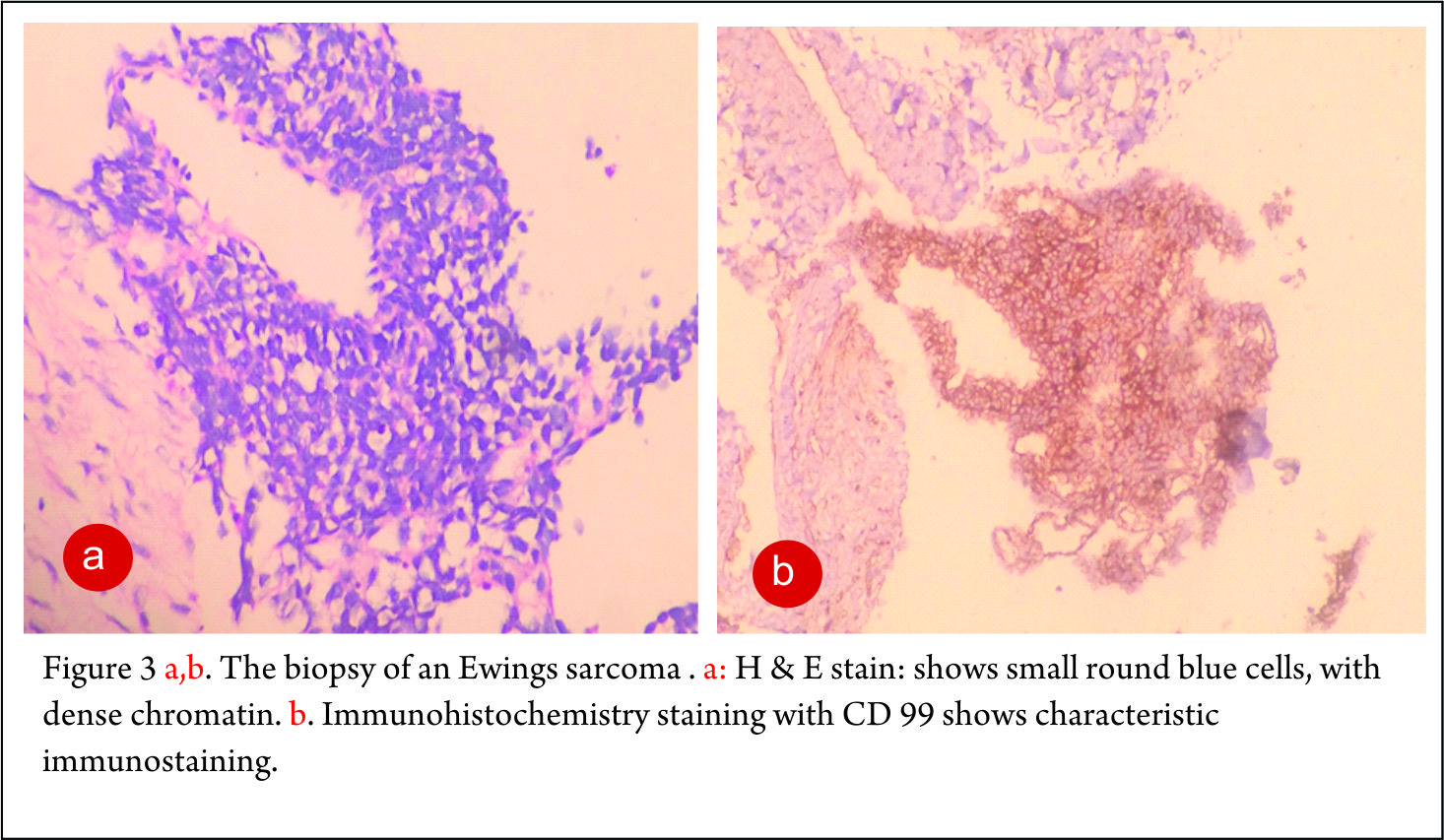
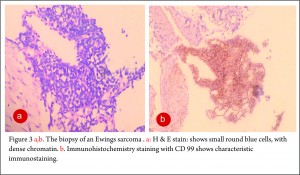
The clinico-radiological suspicion of Ewing sarcoma has to be corroborated by a biopsy and pathological examination before further treatment is commenced. The biopsy of such a lesion is to be done preferably by the orthopaedic oncologist who will be treating the case, at a multi disciplinary cancer centre [3, 11, 13, 14, 15, 16, 17]. Most of the lesions are accurately diagnosed by a needle biopsy. Under the microscope, the tumor is arranged in sheets, nests or clusters of small round blue cells invading the native bone [1]. (Fig 3 a,b). The cells show dense blue chromatin with scanty cytoplasm and the contained glycogen is evident by the P.A.S. (periodic acid-Schiff) stain positivity. Immunohistochemical markers as CD 99 (a mic-2 gene product) and Fli-1 are diagnostic of Ewing sarcoma and are used as confirmatory tools [1,3].
 |
 |
Treatment
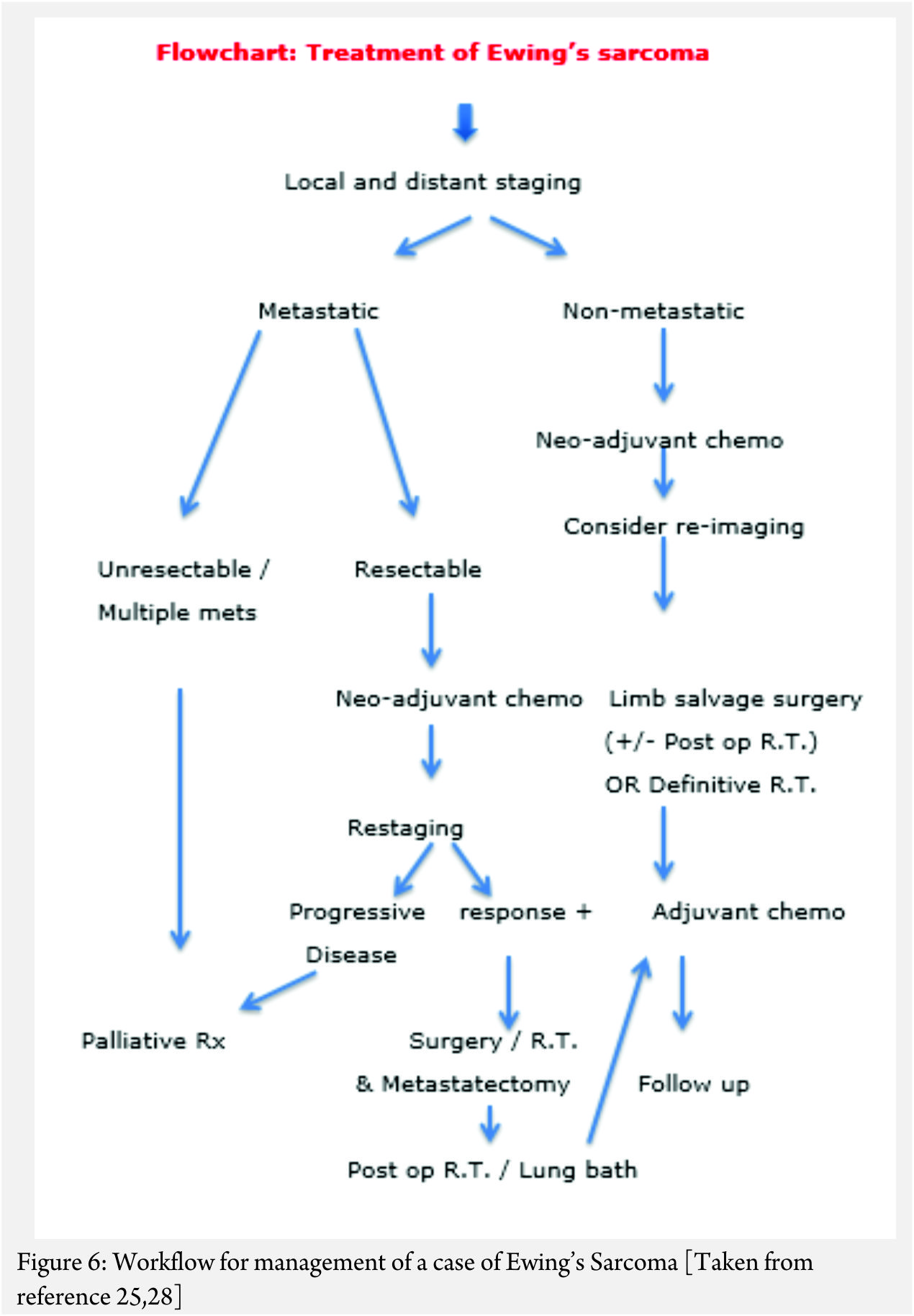
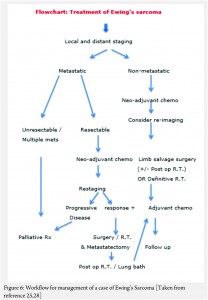
The treatment of ES is handled by a multi disciplinary team comprising of the orthopaedic oncologist, Medical oncologist, Radiation Oncologist, Pathologist and Radiologist [11, 12, 13 , 16, 17, 18]. The patient and the family need to be informed about the clinical results and the expected prognosis and have to participate in the decision making process. Flowcharts of both diagnostic work up and management protocol are provided in figures 5 and 6. The prognosis depends upon the metastatic status of the patient, with the non-metastatic patients having a better outcome [1,3].
The actual management of non-metastatic ES requires neo-adjuvant chemotherapy, followed by surgical resection (if feasible and indicated) followed by post op radiotherapy if necessary (OR definitive local radiotherapy) and then adjuvant (post operative) chemotherapy [1,3] .
In general, the local treatment outcome of an extremity ES is better with surgical wide resection than compared to definitive local radiotherapy alone [18]. In an axially located ES as in pelvis and spine, the decision regarding excision will have to be weighed against the morbidity of the surgery [19]. In a non-metastatic axially located ES, surgery or combined surgery and radiotherapy appears to have an edge over only radiotherapy; the latter being used only in unresectable tumors [20, 21, 22, 23].
The neo adjuvant chemotherapy helps in multiple ways. It is useful in downstaging the local disease, reducing the vascularity, controlling the micro-metastases, sterilizing the satellite lesions in the surrounding zone of hyperemia, helping formation of a thicker capsule, reducing the local edema, healing of pathological fractures and prognosticating outcome of the treatment based on the analysis of percentage necrosis in the tumor. All of these help in making the surgical excision easier and reduce the local recurrence rates [24,25,26,27,28,29,30,31].
The decision regarding limb salvage in a non-metastatic case of Ewings sarcoma is based upon the local extent of the disease. The status of the neuro-vascular bundle and amount of muscles involved by the soft tissue component, determine feasibility of a limb salvage surgery. The only absolute contra indication to a limb salvage surgery would be encasement of a major motor nerve in the extremity and inadequate muscles left after wide excision of the lesion, which would result in a non-functional extremity.
In a case that there are metastases at diagnosis, the decision regarding the approach is based on the number and type of metastases. In a widely metastatic case, only palliative treatment is offered. If there are few pulmonary metastases amenable to excision or are of doubtful significance, the patient is given neo adjuvant chemotherapy and re-staged. The decision regarding treatment is then based on the response to the chemotherapy. If there is progression despite the neo-adjuvant chemotherapy, palliative protocol is followed. If the metastatic lesions have responded to the chemotherapy then the local treatment decision can be taken accordingly with curative intent [25, 28].
The local control rates and the overall survival rates for patients of primary bone sarcomas treated with limb salvage and for those treated by amputation are comparable, with limb salvage surgery carrying better functional outcome [26,28,30,34]. In developing countries, it is worthwhile to offer limb salvage to patients who have a better prognosis, in whom the function of the salvaged extremity is going to be acceptable and for those who are willing to complete the necessary treatment and understand the complications involved.
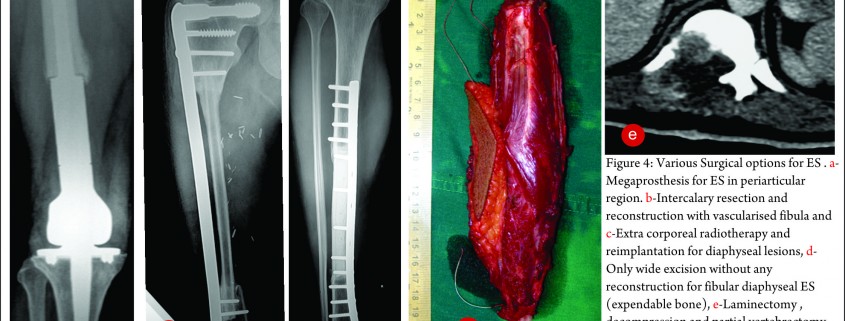
The exact modality of reconstruction after limb salvage is decided by the site of disease, the extent, the patients age [11,34] and expectations and in the developing world, by the socio-economic status of the patient (Fig 4). For periarticular ES, reconstruction can be done by using megaprosthesis [34] or allo-prosthesis composite. This restores the function in the operated extremity fast, shortens the rehabilitation time post operatively, enables early resumption of adjuvant treatment modalities, is a durable option with acceptable complication rate. Arthrodesis can be an alternative to megaprosthetic reconstruction. In cases with diaphyseal involvement, joint sparing inter-calary resections can be done and the defect reconstructed using allograft – live fibula composite or only live vascularised fibula or extra corporeal radiotherapy and reimplantation [35,41]. Rotationplasty is a viable alternative for very young children [36] and in cases of failed limb salvage surgery [37].
The post operative margins of the resected specimen and the percentage necrosis after chemotherapy decide the need for post operative radiotherapy. In cases where the margins are inadequate or the tumor is viable, radiation is used post operatively in order to achieve better control rates [3,21-27,29]. The adjuvant chemo continues in the post operative period. [3, 4, 22, 23, 24, 25, 26, 27, 30, 31, 34].
Patients treated thus need to undergo the prescribed rehabilitation program in order to attain the maximum functional outcome [38]. Functional outcomes in these patients are measured by the Musculo Skeletal Tumor society scoring system (MSTS) or the Toronto extremity salvage score (TESS) [39, 40]. These scores basically reflect the ability of the patient to carry out activities of daily living.

Follow up
The patients are advised to follow up every 3 monthly in the first two years, every six monthly for next three years and annually thereafter. At every visit, radiographs and appropriate staging investigations follow clinical examination [3,25]. Fuchs et al have reported long term complications in 59% percent of patients treated for ES over a average follow up of 25 years [46]. These complications comprised of metastases, local recurrence, secondary malignancies, pathologic fractures, and radiation-associated and chemotherapy-associated morbidities. Hence it is recommended to follow all these patients over a longer period.
Results
In various studies, the overall survival (at 3 or 5 year follow up) for non metastatic ES has been reported to be between 43.5% to 80% [1,23,42-48]. The local recurrence rates are reported to be around 10% to 12.5% [44,48]. In long term follow up of an average of 18 years, Bacci et al have reported overall survival at 5, 10, 15 and 20 years as 57.2%, 49.3%, 44.9% and 38.4% respectively [45]. The poor prognostic indicators in a case of ES are presence of metastases (especially bone and bone marrow metastases), age older than 10 years, a size larger than 200 ml, more central lesions (as in the pelvis or spine), and poor response to chemotherapy [3]. New pharmacological agents and radiotherapeutical modalities are being investigated as discussed in the earlier two articles in the symposium [49,50] and possibility of imporving the survival and quality of life of patients with ES looks promising.
Conclusion
ES is one of the common primary bone malignancies. Appropriate diagnosis, staging, biopsy and treatment at specialized centers is essential for a good outcome. Treatment is multi modal with neoadjuvant and adjuvant chemotherapy, surgery with appropriate margins and radiation in adjuvant or definitive setting; all playing important role in achieving good overall survival rates. Limb salvage surgery in non-metastatic ES is now a norm. The survivors are prone to many long-term complications and need to be followed up for a longer duration. .
References
1. Hameed M. Small Round Cell Tumors Of Bone. Arch Pathol Lab Med. 2007;131:192-204.
2. Rajwanshi A, Srinivas R, Upasana G. Malignant small round cell tumors.J Cytol. 2009;26(1): 1–10.
3. Iwamoto Y. Diagnosis and treatment of Ewing’s sarcoma. Jpn J Clin Oncol. 2007 Feb;37(2):79-89.
4. Bacci G, Ferrari S, Rosito P, Avella M, Barbieri E, Picci P, Battistini A, Brach del Prever A. Ewing’s sarcoma of the bone. Anatomoclinical study of 424 cases. Minerva Pediatr. 1992 Jul-Aug;44(7-8):345-59.
5. Eggli KD, Quiogue T, Moser RP Jr. Ewing’s sarcoma. Radiol Clin North Am. 1993 Mar;31(2):325-37.
6. Bacci G1, Avella M, McDonald D, Toni A, Orlandi M, Campanacci M. Serum lactate dehydrogenase (LDH) as a tumor marker in Ewing’s sarcoma. Tumori. 1988 Dec 31;74(6):649-55.
7. Quartuccio N1, Fox J, Kuk D, Wexler LH, Baldari S, Cistaro A, Schöder H. Pediatric bone sarcoma: diagnostic performance of ¹⁸F-FDG PET/CT versus conventional imaging for initial staging and follow-up. AJR Am J Roentgenol. 2015 Jan;204(1):153-60.
8. Anderson PM. Futility versus utility of marrow assessment in initial Ewing sarcoma staging workup. Pediatr Blood Cancer. 2015 Jan;62(1):1-2.
9. Gerth HU, Juergens KU, Dirksen U, Gerss J, Schober O, Franzius C. Significant benefit of multimodal imaging: PET/CT compared with PET alone in staging and follow-up of patients with Ewing tumors. J Nucl Med. 2007 Dec;48(12):1932-9.
10. Oberlin O1, Bayle C, Hartmann O, Terrier-Lacombe MJ, Lemerle J. Incidence of bone marrow involvement in Ewing’s sarcoma: value of extensive investigation of the bone marrow. Med Pediatr Oncol. 1995 Jun;24(6):343-6.
11. Meyers PA1, Levy AS. Ewing’s sarcoma. Curr Treat Options Oncol. 2000 Aug;1(3):247-57.
12. Exner GU, von Hochstetter AR.Technique and tactics of biopsy including puncture. Z Orthop Ihre Grenzgeb. 1992 Jul-Aug;130(4):272-5.
13. Mavrogenis AF, Angelini A, Vottis C, Palmerini E, Rimondi E, Rossi G, Papagelopoulos PJ, Ruggieri P. State-of-the-art approach for bone sarcomas. Eur J Orthop Surg Traumatol. 2015 Jan;25(1):5-15.
14. Burke NG, Moran CJ, Hurson B, Dudeney S, O’Toole GC. Musculoskeletal oncology training during residency. J Orthop Surg (Hong Kong). 2011 Dec;19(3):350-3.
15. Huang AJ, Kattapuram SV. Musculoskeletal neoplasms: biopsy and intervention. Radiol Clin North Am. 2011 Nov;49(6):1287-305.
16. Scarborough MT. The biopsy. Instr Course Lect. 2004;53:639-44.
17. Bickels J, Jelinek JS, Shmookler BM, Neff RS, Malawer MM. Biopsy of musculoskeletal tumors. Current concepts. Clin Orthop Relat Res. 1999 Nov;(368):212-9.
18. Sudanese A, Toni A, Ciaroni D, Avella M, Dallari D, Picci P, Bacci G, Barbieri E, Mancini A, Campanacci M, et al. The role of surgery in the treatment of localized Ewing’s sarcoma. Chir Organi Mov. 1990 Jul-Sep;75(3):217-30
19. Beadel GP, McLaughlin CE, Aljassir F, Turcotte RE, Isler MH, Ferguson P, Griffin AM, Bell RS, Wunder JS. Iliosacral resection for primary bone tumors: is pelvic reconstruction necessary? Clin Orthop Relat Res. 2005 Sep;438:22-9.
20. Scully SP, Temple HT, O’Keefe RJ, Scarborough MT, Mankin HJ, Gebhardt MC. Role of surgical resection in pelvic Ewing’s sarcoma. J Clin Oncol. 1995 Sep;13(9):2336-41.
21. Carrie C, Mascard E, Gomez F, Habrand JL, Alapetite C, Oberlin O, Moncho V, Hoffstetter S. Nonmetastatic pelvic Ewing sarcoma: report of the French society of pediatric oncology. Med Pediatr Oncol. 1999 Nov;33(5):444-9.
22. Donati D, Yin J, Di Bella C, Colangeli M, Bacci G, Ferrari S, Bertoni F, Barbieri E, Mercuri M. Local and distant control in non-metastatic pelvic Ewing’s sarcoma patients. J Surg Oncol. 2007 Jul 1;96(1):19-25.
23. Biswas B, Rastogi S, Khan SA, Mohanti BK, Sharma DN, Sharma MC, Mridha AR, Bakhshi S. Outcomes and prognostic factors for Ewing-family tumors of the extremities. J Bone Joint Surg Am. 2014 May 21;96(10):841-9.
24. Sciubba DM, Okuno SH, Dekutoski MB, Gokaslan ZL. Ewing and osteogenic sarcoma: evidence for multidisciplinary management. Spine (Phila Pa 1976). 2009 Oct 15;34(22 Suppl):S58-68.
25. ESMO Guidelines Working Group, Saeter G. Ewing’s sarcoma of bone: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2007 Apr;18 Suppl 2:ii79-80.
26. Bacci G, Balladelli A, Forni C, Ferrari S, Longhi A, Bacchini P, Alberghini M, Fabbri N, Benassi M, Briccoli A, Picci P. Adjuvant and neoadjuvant chemotherapy for Ewing sarcoma family tumors in patients aged between 40 and 60: report of 35 cases and comparison of results with 586 younger patients treated with the same protocols in the same years. Cancer. 2007 Feb 15;109(4):780-6.
27. Sluga M, Windhager R, Lang S, Heinzl H, Krepler P, Mittermayer F, Dominkus M, Zoubek A, Kotz R. A long-term review of the treatment of patients with Ewing’s sarcoma in one institution. Eur J Surg Oncol. 2001 Sep;27(6):569-73.
28. Paulussen M, Ahrens S, Craft AW, Dunst J, Fröhlich B, Jabar S, Rübe C, Winkelmann W, Wissing S, Zoubek A, Jürgens H. Ewing’s tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing’s Sarcoma Studies patients. J Clin Oncol. 1998 Sep;16(9):3044-52.
29. Picci P, Rougraff BT, Bacci G, Neff JR, Sangiorgi L, Cazzola A, Baldini N, Ferrari S, Mercuri M, Ruggieri P, et al. Prognostic significance of histopathologic response to chemotherapy in nonmetastatic Ewing’s sarcoma of the extremities. J Clin Oncol. 1993 Sep;11(9):1763-9.
30. Bacci G, Ferrari S, Avella M, Barbieri E, Picci P, Casadei R, Rosito P, Neri S, Capanna R, Battistini A, et al. Non-metastatic Ewing’s sarcoma: results in 98 patients treated with neoadjuvant chemotherapy. Ital J Orthop Traumatol. 1991 Dec;17(4):449-65.
31. Cagnano R, Avella M, Rosito P, Ciaroni D, Ferraro A, Di Scioscio M, Putti C, Bacci G. Neoadjuvant chemotherapy for localized Ewing’s sarcoma. Preliminary results of a new protocol which uses surgery alone or followed by radiotherapy for local control. J Chemother. 1989 Jul;1(4 Suppl):1248-9.
32. Sluga M, Windhager R, Lang S, Heinzl H, Krepler P, Mittermayer F, Dominkus M, Zoubek A, Kotz R.The role of surgery and resection margins in the treatment of Ewing’s sarcoma. Clin Orthop Relat Res. 2001 Nov;(392):394-9.
33. Hobusch GM, Lang N, Schuh R, Windhager R, Hofstaetter JG. Do patients with ewing’s sarcoma continue with sports activities after limb salvage surgery of the lower extremity? Clin Orthop Relat Res. 2015 Mar;473(3):839-46.
34. Dai X, Ma W, He X, Jha RK. Review of therapeutic strategies for osteosarcoma, chondrosarcoma, and Ewing’s sarcoma. Med Sci Monit. 2011 Aug;17(8):RA177-190.
35. Poffyn B, Sys G, Mulliez A, Van Maele G, Van Hoorebeke L, Forsyth R, Uyttendaele D. Extracorporeally irradiated autografts for the treatment of bone tumours: tips and tricks. Int Orthop. 2011 Jun;35(6):889-95.
36. Hardes J, Gosheger G, Vachtsevanos L, Hoffmann C, Ahrens H, Winkelmann W. Rotationplasty type BI versus type BIIIa in children under the age of ten years. Should the knee be preserved? J Bone Joint Surg Br. 2005 Mar;87(3):395-400.
37. Ramseier LE, Dumont CE, Exner GU. Rotationplasty (Borggreve/Van Nes and modifications) as an alternative to amputation in failed reconstructions after resection of tumours around the knee joint. Scand J Plast Reconstr Surg Hand Surg. 2008;42(4):199-201
38. Shehadeh A, El Dahleh M, Salem A, Sarhan Y, Sultan I, Henshaw RM, Aboulafia AJ. Standardization of rehabilitation after limb salvage surgery for sarcomas improves patients’ outcome. Hematol Oncol Stem Cell Ther. 2013 Sep-Dec;6(3-4):105-11
39. Cassidy RJ, Indelicato DJ, Gibbs CP, Scarborough MT, Morris CG, Zlotecki RA. Function Preservation After Conservative Resection and Radiotherapy for Soft-tissue Sarcoma of the Distal Extremity: Utility and Application of the Toronto Extremity Salvage Score (TESS). Am J Clin Oncol. 2014 Jul 17.
40. P. U. Tunn & D. Pomraenke & U. Goerling & P. Hohenberger. Functional outcome after endoprosthetic limb-salvage therapy of primary bone tumours—a comparative analysis using the MSTS score, the TESS and the RNL index. International Orthopaedics (SICOT) (2008) 32:619–625.
41. Puri A, Gulia A, Jambhekar N, Laskar S. The outcome of the treatment of diaphyseal primary bone sarcoma by resection, irradiation and re-implantation of the host bone: extracorporeal irradiation as an option for reconstruction in diaphyseal bone sarcomas. J Bone Joint Surg Br. 2012 Jul;94(7):982-8
42. Bacci G, Palmerini E, Staals EL, Longhi A, Barbieri E, Alberghini M, Ferrari S Ewing’s sarcoma family tumors of the humerus: outcome of patients treated with radiotherapy, surgery or surgery and adjuvant radiotherapy. Radiother Oncol. 2009 Nov;93(2):383-7.
43. La TH, Meyers PA, Wexler LH, Alektiar KM, Healey JH, Laquaglia MP, Boland PJ, Wolden SL. Radiation therapy for Ewing’s sarcoma: results from Memorial Sloan-Kettering in the modern era. Int J Radiat Oncol Biol Phys. 2006 Feb 1;64(2):544-50.
44. Krasin MJ1, Davidoff AM, Rodriguez-Galindo C, Billups CA, Fuller CE, Neel MD, Merchant TE. Definitive surgery and multiagent systemic therapy for patients with localized Ewing sarcoma family of tumors: local outcome and prognostic factors. Cancer. 2005 Jul 15;104(2):367-73.
45. Bacci G1, Forni C, Longhi A, Ferrari S, Donati D, De Paolis M, Barbieri E, Pignotti E, Rosito P, Versari M. Long-term outcome for patients with non-metastatic Ewing’s sarcoma treated with adjuvant and neoadjuvant chemotherapies. 402 patients treated at Rizzoli between 1972 and 1992. Eur J Cancer. 2004 Jan;40(1):73-83.
46. Fuchs B1, Valenzuela RG, Inwards C, Sim FH, Rock MG. Complications in long-term survivors of Ewing sarcoma. Cancer. 2003 Dec 15;98(12):2687-92.
47. Marcus Jr RB, Berrey BH, Graham-Pole J, Mendenhall NP, Scarborough MT. The treatment of Ewing’s sarcoma of bone at the University of Florida: 1969 to 1998. Clin Orthop Relat Res. 2002 Apr;(397):290-7.
48. Elomaa I1, Blomqvist CP, Saeter G, Akerman M, Stenwig E, Wiebe T, Björk O, Alvegård TA. Five-year results in Ewing’s sarcoma. The Scandinavian Sarcoma Group experience with the SSG IX protocol. Eur J Cancer. 2000 May;36(7):875-80.
49. Irukulla MM, Joseph DM. Management of Ewing Sarcoma:Current Management and the Role of Radiation Therapy. Journal of Bone and Soft Tissue Tumors May-Aug 2015; 1(1):4-6
50.Valvi S & Kellie SJ. Ewing Sarcoma: Focus on Medical Management. Journal of Bone and Soft Tissue Tumors May-Aug 2015; 1(1):4-6.
| How to Cite this article: Panchwagh Y. Ewing Sarcoma: Focus on Surgical Management. Journal of Bone and Soft Tissue Tumors May-Aug 2015;1(1):23-28. |
 - Dr.Yogesh Panchwagh
|
Like this:
Like Loading...