Vol 1 | Issue 1 | May – August 2015 | page:1-2 | Santosh Valvi, Stewart J Kellie
Author: Santosh Valvi [1,2*], Stewart J Kellie [3,4]
[1]Kids Cancer Centre, Sydney Children’s Hospital, Randwick 2031, New South Wales, Australia
[2] Children’s Cancer Institute Australia, Lowy Cancer Research Centre, University of New South Wales, Randwick 2031, New South Wales, Australia
[3] Oncology Unit, The Children’s Hospital at Westmead, Westmead 2145, New South Wales, Australia
[4] Discipline of Paediatrics and Child Health, Faculty of Medicine, University of Sydney, Westmead 2145, New South Wales, Australia
Address of Correspondence
Dr. Santosh Valvi FRACP
Kids Cancer Centre, Sydney Children’s Hospital, Randwick 2031, New South Wales, Australia
Email: santosh.valvi@health.nsw.gov.au
Abstract
The management of Ewing sarcoma has evolved over the last few decades with successive improvement in survival rates. Multidisciplinary management is the key to successful outcomes. Dose intensity of chemotherapy is of vital importance. Local control can be effectively achieved with surgery, radiation therapy or a combination of the two. The choice of appropriate local therapy should be individualized and depends on various factors such as site, size, respectability, expected morbidity, long term effects etc. Metastatic disease remains a significant challenge and optimal therapeutic strategies still need to be defined. Current management and the role of radiation therapy in Ewing sarcoma are reviewed.
Keywords: Ewing sarcoma, radiation therapy, management
Introduction
In 1921, James Ewing reported a group of primary radiosensitive tumors as diffuse endothelioma of bone, believing they arose from the blood vessels of bone tissue [1]. A few years later the noted Boston surgeon, Ernest Codman, referred to this new entity as Ewing sarcoma (EWS) [2]. EWS, a rare malignancy with a strong pediatric predilection, typically presents as a bone tumor [3]. It is the second most common primary malignant bone tumor in children and young adults, following osteosarcoma and accounts for approximately 3% of all childhood malignancies [4].
Epidemiology
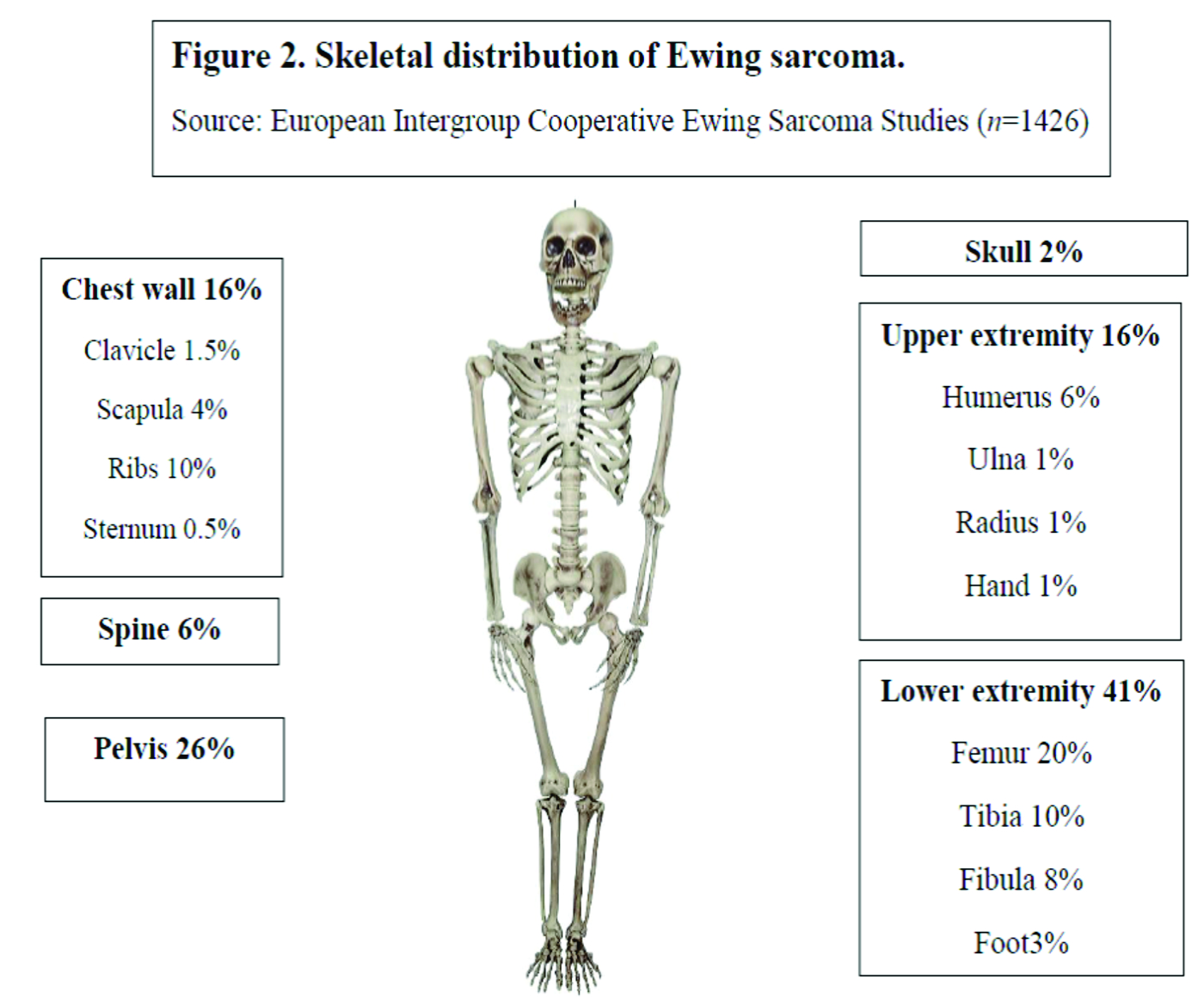
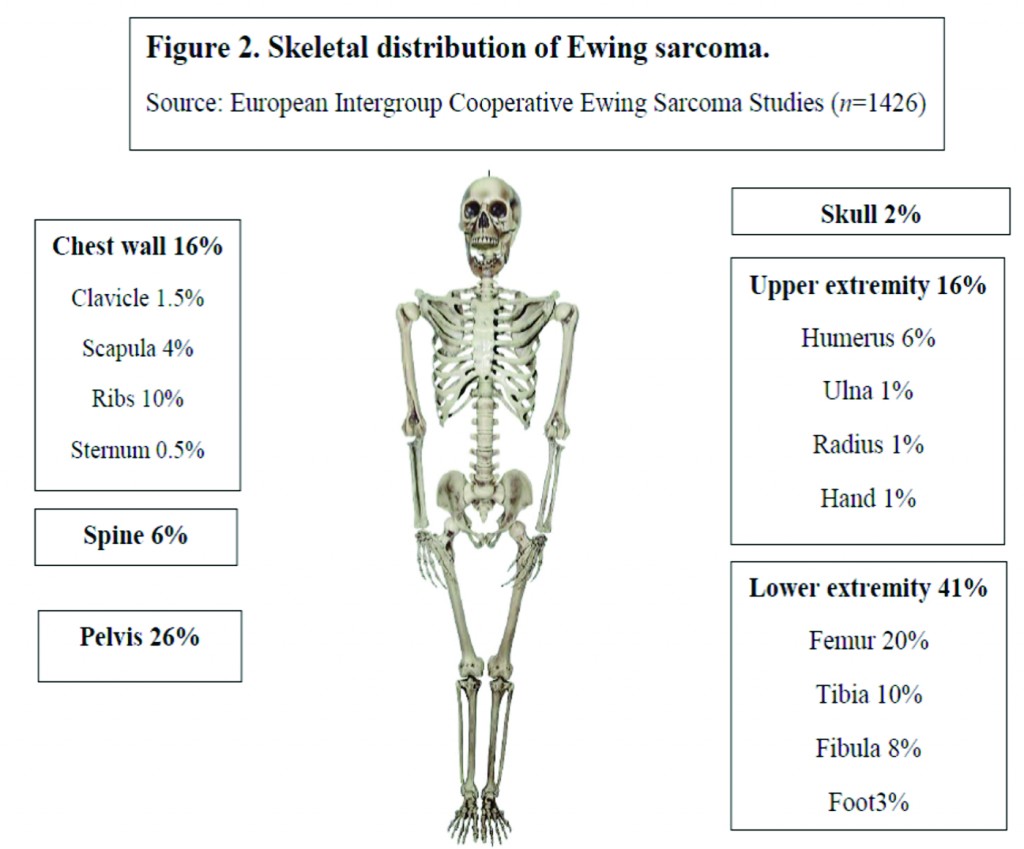
Over the last 30 years, the incidence of EWS has remained unchanged at around 3 cases per million per year [5]. With a median age of 15 years, it most commonly occurs in the second decade of life (Fig 1) [6]. There is a slight male predilection (male: female 1.2:1) and Caucasians are much more frequently affected than Asians and Africans [7,8]. Lower extremities are the most common site of bone disease (43%) while extraosseous primary tumors mostly occur in the trunk (32%) (Fig 2). Metastatic disease is present at diagnosis in about 20-25% of patients and affects the lungs, other bones or multiple systems [5,9].
Biology & Pathology
The World Health Organisation (WHO) classification uses EWS/primitive neuroectodermal tumor (PNET) as an inclusive term which encompasses classic EWS, Askin tumor of the thoracic wall, Ewing tumor, peripheral neuroepithelioma, peripheral neuroblastoma, Ewing family of tumors and Ewing sarcoma family of tumors [10]. EWS is derived from a primordial bone marrow-derived mesenchymal stem cell [11,12]. Histologically, EWS is characterised by a monotonous population of small round blue cells with a low mitotic activity of 15-20%. Cytoplasmic glycogen is abundant which gives periodic acid-Schiff (PAS) positivity [13]. The MIC2 gene product, CD99, a surface membrane glycoprotein is overexpressed [14] but it is not specific for EWS. Neural differentiation is evident in the form of positive vimentin in approximately one third of cases.
A reciprocal chromosomal translocation involving the EWSR1 gene on chromosome 22 band q12 combined with any of a number of partner chromosomes is pathognomonic of the diagnosis of EWS. The breakpoint was first cloned in the 1990s [12,15]. Although abnormalities of chromosome 11 are involved in 95% of cases [16], the translocation may involve chromosomes 21, 7 and 17 uncommonly [17,18]. The fusion protein resulting from this chromosomal rearrangement is a potent transcriptional factor which inappropriately activates the target genes, thereby exerting the oncogenic activity.
Other numerical and structural alterations seen in EWS are gains of chromosomes 2, 5, 8, 9, 12, and 15; deletions on the short arm of chromosome 6; the nonreciprocal translocation t(1;16)(q12;q11.2); and trisomy 20 [19,20].
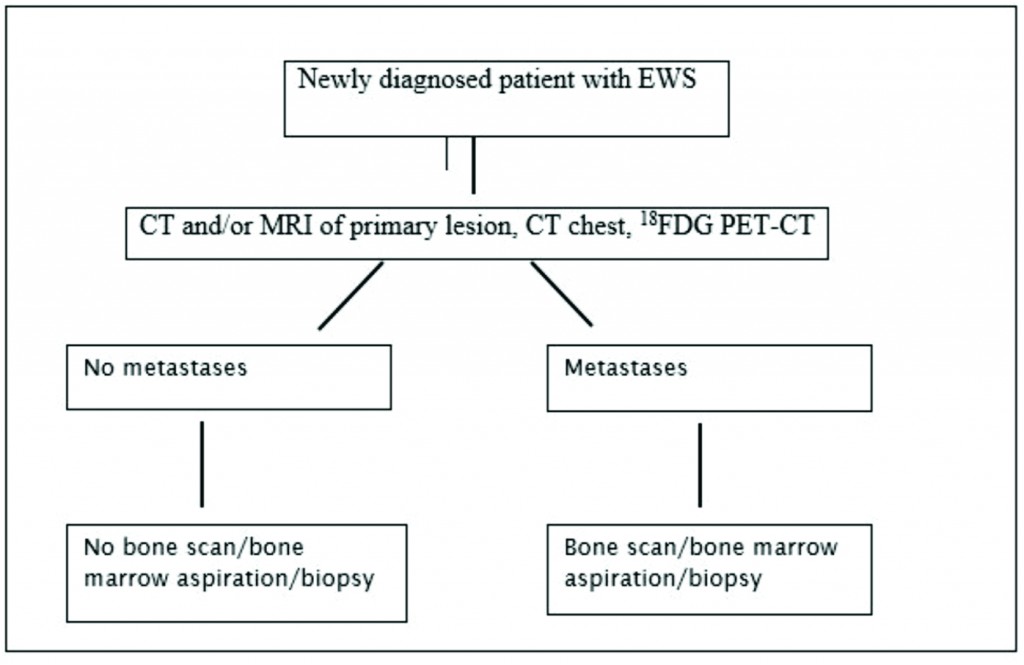
- Figure 1: Investigation Workflow for a newly diagnosed Patient with EWS
Staging
EWS is defined by clinical and imaging techniques as localized when there is no spread beyond the primary site or metastatic when the tumor has disseminated to distant organs. Of all imaging modalities, 18FDG PET-CT has the highest specificity (96%) and sensitivity (92%) [21] and is superior to the traditionally used 99mTc-MDP bone scan for detection of bone metastases except for skull lesions [22]. Current recommendations for staging work-up include CT and/or MRI of the primary tumor, chest CT to detect lung metastases and 18FDG PET-CT for identification of distant metastases [23]. As bone marrow involvement is an independent risk factor [24], marrow biopsy has been an integral part of the initial work-up and is still recommended in ongoing clinical trials [25] (26). But recent studies have questioned the utility of bone marrow biopsy in localized [22,27] and metastatic disease [23].
Prognosis
The 5 year survival rate for EWS was less than 10% before the advent of modern chemotherapy [28,29]. Currently, the survival rates are 70% for the patients with localized disease [30] and 30% for the patients with metastatic disease [9]. Among patients with refractory or recurrent disease, fewer than 20% of patients can expect to be long term survivors [31,32].
The presence of metastatic disease at diagnosis remains the most important adverse prognostic factor in EWS [33,34,35,36]. In patients with metastatic disease the site(s) of metastases can have an impact on the outcome. Patients with only lung metastases fare better (event free survival, EFS 29% to 52%) than patients with bone and/or bone marrow involvement (EFS 19%) [37,38] or combined bones and lungs involvement (EFS 8%) (34). Unilateral lung involvement has a better outcome compared with bilateral lung lesions [39].
Younger age (<15 years old) [5,40,41], female gender [42], tumor site (distal extremity better than proximal extremity and pelvis) [9], tumor size (volume less than 200 ml and single dimension less than 8 cm) [43], normal serum lactate dehydrogenase (LDH) levels at diagnosis [44], and decreased metabolic activity on 18FDG PET scan after presurgical chemotherapy [45,46] are associated with a more favourable prognosis.
Complex karyotypic abnormalities or chromosome number less than 50 in tumor cells at diagnosis [19], detection of fusion transcripts by polymerase chain reaction (PCR) in morphologically normal bone marrow [47], p53 protein overexpression, Ki67 expression, loss of 16q [48,49], overexpression of microsomal glutathione S-transferase (associated with doxorubicin resistance [50] may be associated with inferior outcome. Patients with secondary Ewing sarcoma [51] or with a poor response to presurgical chemotherapy [52,53] and patients relapsing less than two years after diagnosis (early) have a poorer prognosis [54].
Treatment options
Chemotherapy for a total of 10-12 months before and after local control is common practice [33,55]. Initial chemotherapy aims to shrink the tumor to increase to probability of effective local control. Alkylating agents, mainly ifosfamide and cyclophosphamide and anthracyclines form the chemotherapeutic backbone Etoposide, vincristine and actinomycin-D make up the remainder of the four-to five-drug combination chemotherapy.
Chemotherapy for newly diagnosed patients:
Clinical trials in the early years (pre-1990)
Before 1960s, radiation therapy and surgery were used for the treatment of EWS which provided adequate control of the primary disease but patients invariably died of metastatic disease [56]. Chemotherapy was added based on the hypothesis that, in most cases of apparently localized disease, tumor cells were already disseminated without clinical manifestations. Single chemotherapy agents including cyclophosphamide [57,58,59], vincristine [60], daunorubicin [61] and actinomycin-D [62] were trialled in 1960s with promising results.
From two- to as many as six-drug combinations have been used in various randomized and non-randomized trials for the treatment of EWS. Hustu et al [63] used a first ever combination with vincristine and cyclophosphamide with 80% overall survival. In Europe, the French Society of Pediatric Oncology (SFOP) [64,65,66], the United Kingdom Children’s Cancer Study Group (UKCCSG) [35,67], the Scandinavian Study Group (SSG) [68, 69] and the German/Austrian Cooperative Ewing Sarcoma Study Group (CESS) [70,71] performed early clinical trials. Subsequently, the European Intergroup Cooperative Ewing Sarcoma Study group (EICESS) and the European Ewing Tumor Working Initiative of National Groups (EURO-EWING) continued the trials. In the United States, initially the Intergroup Ewing Sarcoma Study (IESS) group [72,73,74], the Children’s Cancer Group (CCG), the Pediatric Oncology Group (POG) and subsequently the Children’s Oncology Group (COG) conducted trials for EWS.
Four-drug combination chemotherapy including vincristine, actinomycin-D, cyclophosphamide and doxorubicin was universally accepted for the treatment by the early 1980s [75] with survival rates between 36-60%. Ifosfamide and etoposide were identified as effective single agents [76,77] and subsequent studies established a survival benefit of their addition to VACD [78]. National Cancer Institute protocol INT0091 was a randomized trial conducted by the Children’s Cancer Group (CCG) and Pediatric Oncology Group (POG) from 1988 through 1992. Patients were assigned to receive VACD or VACD plus ifosfamide and etoposide (VACD-IE). In patients without metastatic disease, the five-year EFS for the VACD group was 54% while the same for the VACD-IE group was 69%. These results established VACD-IE as the gold standard for the treatment of localised Ewing sarcoma [30].
Clinical trials for standard risk (SR) and high risk (HR) EWS since 1990
The disease risk stratification into SR and HR has varied depending on the trial but in general SR means localized small tumors (<200 mL), or tumors with a good histological response to preoperative chemotherapy (<10% cells). HR tumors include metastatic tumors, or large localized tumors (>200 mL).
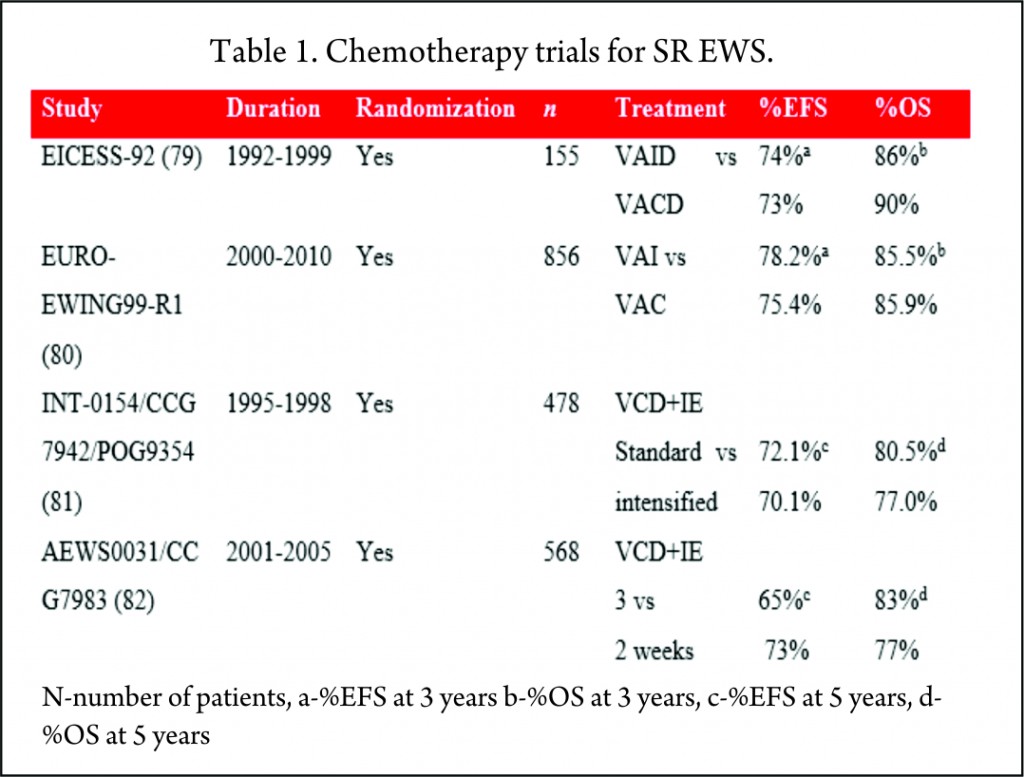
The trials for SR EWS have tried to address the important questions like the superiority of one alkylating agent over the other (cyclophosphamide and ifosfamide) and survival advantage by dose intensification or addition newer chemotherapy agents.
Cyclophosphamide vs Ifosfamide
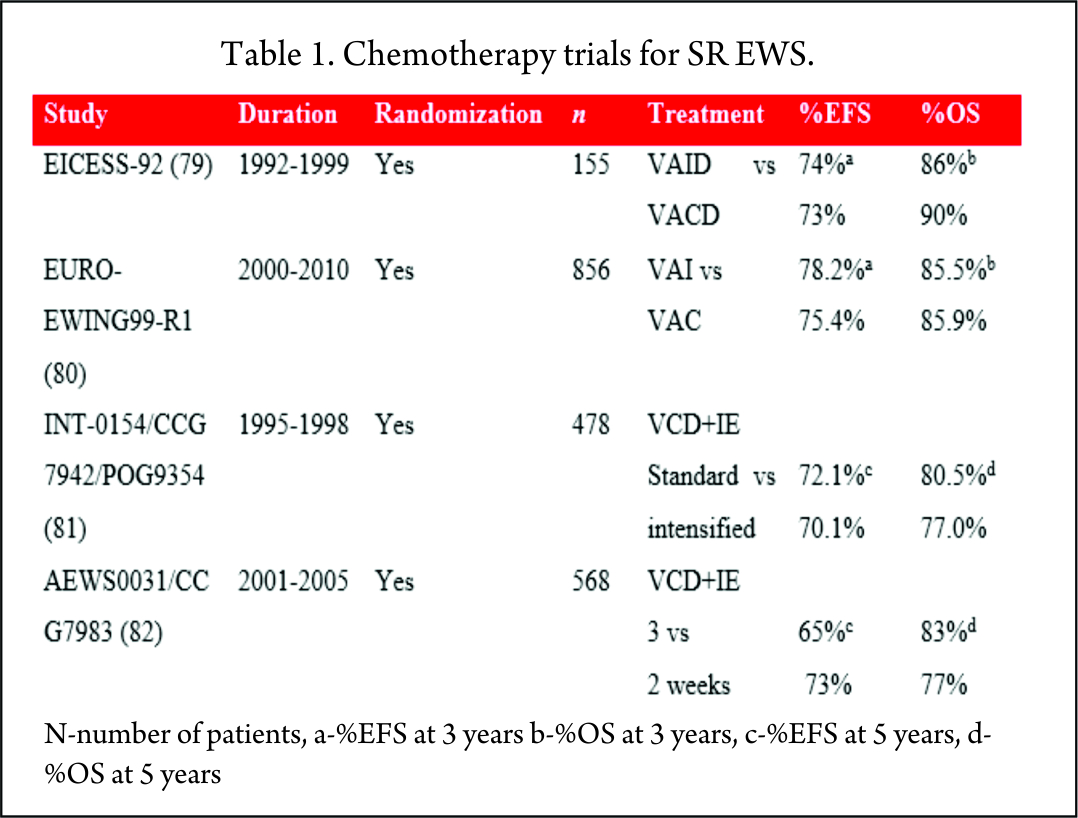
Historically, cyclophosphamide was used for the treatment of EWS. Promising results were seen with ifosfamide in relapsed patients who did not respond to cyclophosphamide [83]. It was postulated that 9 g/m2 of ifosfamide was equimyelotoxic to 2.1 g/m2 of cyclophosphamide [84]. With the potential for less myelotoxicity and high-dose administration, cyclophosphamide was replaced with ifosfamide in the 1980s. But the results of these non-randomized, single-arm studies were mixed, with one study showing no benefit [66] while others proving superiority of ifosfamide over cyclophosphamide [71,67,69]. With this uncertainty of greater efficacy and long-term renal tubular damage with the cumulative dose of ifosfamide [85], its role in the consolidation treatment of EWS was debated. Two large randomized trials, EICESS-92 [79] and its successor Euro-Ewing99-R1 [80] investigated if cyclophosphamide can replace ifosfamide in the consolidation treatment of standard-risk EWS. The results of these studies confirmed that both the drugs had similar efficacy and though cyclophosphamide was associated with more haematological toxicity, the incidence of renal toxicity was much less as compared to ifosfamide. But the question of superiority of one drug over the other is far from resolved and needs further investigation in light of their efficacy to improve the survival [75].
Standard dose vs dose intensification
To improve the outcome, intensification of chemotherapy drug doses was investigated. One way of achieving dose intensification is by escalating the doses of chemotherapy agents while keeping the interval stable. National Cancer Institute protocol INT0154 used VDC+IE chemotherapy and randomized patients to standard (17 cycles over 48 weeks) or intensified (11 cycles over 30 weeks) arms. This study showed no improvement in the outcome of patients with nonmetastatic disease by dose escalation of alkylating agents (81) which was in contrast to an earlier similar study, IESS-II [74].
AEWS0031 trial investigated the feasibility of dose intensification by interval compression (increased dose density) in patients with localized disease [82]. Patients treated every two weeks (intensified arm) had an improved five-year EFS (73%) compared with the standard arm group receiving chemotherapy every 3 weeks (65%) with no increase in toxicity. Due to its superiority, interval compression is used in many ongoing trials.
The Children’s Oncology Group is currently conducting a phase III randomized trial of adding vincristine, topotecan and cyclophosphamide to standard chemotherapy for patients with localized EWS in an attempt to improve the outcome further [25].
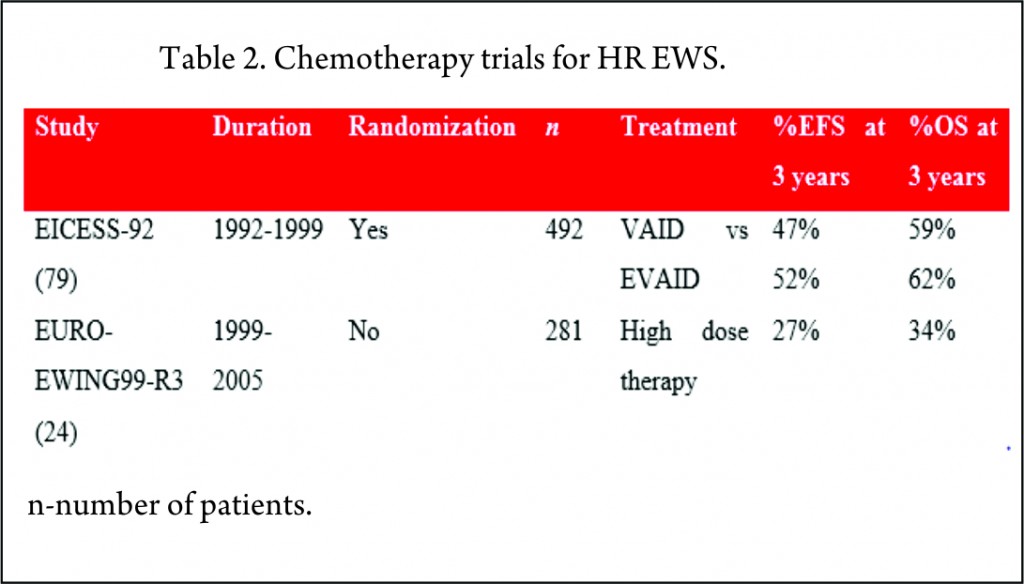
The EICESS-92 study recruited 492 high risk patients of which 157 had metastatic disease at diagnosis. These patients were randomized to receive either VAID or etoposide in addition to VAID (EVAID). Although there was evidence that etoposide had a more pronounced effect in localized HR group, there was no benefit for the patients with metastatic disease with a three-year EFS of 30% [79].
The EURO-EWING99-R3 study enrolled 281 patients with primary disseminated multifocal EWS. 169 patients received the high dose therapy (HDT)/stem cell transplant (SCT) post completion of chemotherapy and local therapy. 3-year EFS for whole cohort was 27% and for patients receiving HDT was 37% [24].
Local therapy
The goal of local therapy is to maximize the local control with minimal morbidity. Surgery and radiation therapy are the two local control modalities employed for EWS. No randomized trials have compared these and as such their relative roles remain controversial [13].
Surgical resection provides information about the amount of tumor necrosis and may be less morbid in the younger patients. Radiation therapy is also associated with the development of second malignant neoplasms in a dose and time dependent manner [86]. A retrospective analysis of patients treated on three consecutive clinical trials for localized EWS showed that the risk of local failure was greater for patients receiving definitive radiotherapy but the EFS and OS were comparable for both surgery and radiation as local control modalities [87]. Microscopically complete surgical resection of localised disease remains the goal of neoadjuvant (or upfront) chemotherapy. Large bone defects after the surgery may be reconstructed using autogenous or allogenic bone grafts and endoprosthetic replacements [13]. Radiation therapy may be used as the main modality of primary disease control in patients with axial or unresectable primary disease. Careful consideration about the use of radiation, dose and volume is required, particularly in younger patients.
In patients with lung metastases, upfront whole-lung radiation may be used irrespective of the radiographic response following chemotherapy [88]. The results of the recently concluded Euro-EWING99 R2 pulmonary (AEWS0331) study which compared the HDT and peripheral blood stem cell (PBSC) rescue with the standard chemotherapy and whole lung irradiation are awaited. A multivariate analysis of the R3 arm of this trial including patients with metastatic disease emphasized the importance of aggressive local control of primary and metastatic sites. The EFS was higher with combined surgery and radiation compared to either modality alone or no local control [89].
High-dose therapy (HDT) and stem cell transplantation (SCT)
Despite advances in multimodal therapy of EWS, there remains a group of patients with high risk of treatment failure. These are primarily the patients with metastatic disease or with extensive unresectable localized disease and patients with a poor response to chemotherapy. This group has a poor 20%-30% disease free survival (DFS) [90,91]. Although conventional chemotherapy regimens induce remission, patients with metastatic disease relapse after a median of one to two years after completion of therapy owing to minimal residual or metastatic disease (MRD/MMD). In the 1980s trials investigating the role of SCT to consolidate remission by reduction of MRD/MMD began. The results of the initial National Cancer Institute (NCI) studies investigating total body irradiation (TBI) with autologous bone marrow transplant (ABMT) showed no improvement in survival [92]. Since then multiple reports have been published of consolidation using HDT followed by SCT but its role in the treatment of EWS has yet to be conclusively determined [93].
Melphalan vs busulfan-based conditioning regimens
Response to melphalan-based HDT has been variable. Some studies showed no additional benefit with poor survival rates between 5%-27% [34,90,94,95] while others [96,97,98] reported improved survival rates of 45%-50%. As use of high-dose busulfan combined with melphalan or other agents has shown promising results with survival rates between 36%-60% [99,100,101,102,103,104], these regimens have been widely used in high-risk patients.
Role of total-body irradiation (TBI)
Use of TBI during the consolidation phase had no survival advantage but increased the incidence of toxicity [92,94]. Two Meta European Intergroup Cooperative Ewing Sarcoma Studies (MetaEICESS) assessed the role of TBI in consolidation treatment. Patients received systemic consolidation in the form of hyperfractionated TBI with melphalan/etoposide in the first HyperME study or two times the melphalan/etoposide in the second TandemME study. EFS were similar in both studies while TBI containing regimen was associated with a higher incidence of toxicity [105]. In conclusion, although EWS is a radiosensitive tumor, there is limited role of TBI in its treatment because of poor efficacy and increased toxicity.
Autologous vs allogenic BMT
Allogeneic transplant may overcome the concerns with tumor cell contamination of stem cell products during autologous transplant [106] and have a potential of graft-versus-tumor (GVT) effect with improved survival. A retrospective analysis of the MetaEICESS study data showed that the EFS was 25% after autologous and 20% after allogeneic transplant [54]. As there was increased incidence of toxicity and no evidence of GVT effect after allogeneic transplant, there seems to be no advantage of allogeneic over autologous transplant.
Chemotherapy for recurrent EWS
Although around 80% of relapses occur within 2 years of initial diagnosis [107], late relapses occurring more than five years from the initial diagnosis are more common in EWS than any other pediatric solid tumors. The Childhood Cancer Survivor Study (108) retrospectively assessed more than 12,700 childhood cancer survivors and concluded that survivors with EWS were at a higher risk of late recurrence, 5-20 years after the diagnosis, than survivors with acute lymphoblastic leukemia. Time to relapse is an important prognostic factor with recurrences occurring within two years of initial diagnosis having worse five-year survival of 7% compared to 30% for patients relapsing after two years [32,107]. Number of recurrences also impacts the outcome with multiple metastatic recurrences having worse prognosis than isolated local or metastatic recurrence [107]. There is no established treatment for these patients and the preferred approach is to combine multi-agent chemotherapy with local modality of surgery and/or radiotherapy [109,110].
High dose Ifosfamide alone [111] or with carboplatin and etoposide (ICE) has been commonly used with survival rates between 29%-33% [112,113]. Cyclophosphamide and topotecan combination achieved response rates of 23%-44% with low toxicity and an added advantage of outpatient administration [114,115] but with a small median duration of response of 8 months [116]. Response rates of 29% to 68% and median time to progression of 3 to 8.5 months were seen with irinotecan and temozolomide [117,118,119,120]. Diarrhea was a troublesome complication which was managed effectively with oral cephalosporins. The combination was otherwise well tolerated. Although gemcitabine and docetaxel showed activity in one study [121], the results were not confirmed by subsequent studies. [122].
In case of recurrent EWS, the addition of HDT to salvage regimens is controversial. Some studies showed a good response in specific groups of patients who responded to relapse therapy and underwent HDT with OS rates of 53 to 66% [123,124], but most of the reports indicate HDT does not improve prognosis [54,125,126].
Targeted therapy for EWS
Tyrosine kinase (TK) inhibitors
TKs are important modulators of growth factor signaling and play a critical role in tumor growth. TK inhibitors are used alone or in combination with conventional chemotherapy agents in treatment of various cancers (127). A number of TK inhibitors have been tried in EWS with variable response.
Insulin-like growth factor 1 receptor (IGF1R) inhibitors
IGF1R is necessary for growth and development of normal as well as cancer cells [128]. With promising pre-clinical results showing IGF1R inhibition in EWS cell lines and xenografts [129], more than 25 agents inhibiting IGF1R are currently under investigation [130].
IGF1R monoclonal antibodies including R1507 (131), figitumumab [132], ganitumab (AMG479) [133], cixutumumab [134,135], and robatumumab (SCH-717454) [136] have shown activity in early phase clinical trials with response rates ranging from 6-14% and a favourable safety profile. But the results of the phase II studies were less impressive compared with the promising preclinical and early clinical data [137]. Small-molecule inhibitors of IGF1R such as GSK1838705A [138], GSK1904529A [139], BMS-754807 [140], and INSM-18 [141] are also in preclinical and clinical development.
Phase II clinical trials of imatinib, a TK inhibitor of the BCR-ABL fusion protein [142,143,144] and dasatinib, a multitargeted TK inhibitor [145] showed no efficacy in EWS.
Biologic agents
Angiogenesis inhibitors
Neovascularization plays a critical role in the pathogenesis of EWS [146] and targeting vascular endothelial growth factor (VEGF) may interfere with vasculogenesis, providing a novel therapeutic approach [147]. A phase I study [148] and a randomized phase II trial [149] conducted by the Children’s Oncology Group have shown the feasibility and tolerability of bevacizumab in EWS patients. Another phase II study investigated the role of vinblastine and celecoxib as angiogenesis inhibitors in combination with the standard chemotherapy (150). Although the feasiblity of this combination was established, there were significant pulmonary and bladder toxicities.
Histone deacetylase (HDAC) inhibitors
HDAC inhibition suppresses EWS-FLI1 expression and may represent a novel therapeutic target for EWS (151).
Mammalian target of rapamycin (mTOR) inhibitors
MTOR is a serine/threonine kinase with critical role in protein synthesis, cell growth and proliferation regulation. mTOR inhibitors have shown activity in preclinical models. A phase I study of temsirolimus, irinotecan and temozolomide demonstrated efficacy and tolerability [152]. But another phase II study of temsirolimus with cixutumumab did not show any objective response despite the encouraging preclinical data [153]. Ridaforolimus was associated with a statistically significant but clinically small benefit on PFS [154].
Aurora A kinase inhibitors
Although alisertib (MLN8237), an Aurora A kinase inhibitor produced promising results in the Pediatric Preclinical Testing Program [155], a recently concluded Children’s Oncology Group phase II trial failed to establish its efficacy in EWS [156].
Hedgehog pathway modulation
Arsenic trioxide was effective in inhibiting EWS growth in preclinical cell culture models by targeting p38(MAPK) and c-Jun N-terminal kinase [157]. These observations warrant further investigation.
Bisphosphonates
Zoledronic acid acts by inducing apoptosis by upregulating osteoprotegerin which was the basis of activity seen in EWS pre-clinical models [158,159]. However, confirmatory clinical trials have not been performed.
Immune therapy
Interleukin-15-activated natural killer (NK) cells combined with HDAC inhibitors improve immune recognition of therapy-sensitive and –resistant EWS and sensitize for NK cell cytotoxicity [160]. Allogenic NK cells have shown activity against EWS cells on their own [161].
EWS-FLI1 targeting
Targeting the EWS-FLI1 fusion protein or its key signalling pathway is another attractive approach [162]. YK-4279, a small molecule inhibitor of EWS-FLI1 protein activity [163,164], mithramycin, a chemotherapy drug [165] and midostaurin (PKC412), a multikinase inhibitor [166] have shown activity in preclinical models.
Conclusion
Many advances have been made in the management of EWS since its first description almost 100 years ago. Molecular and imaging techniques are progressing at a rapid pace allowing for newer insights into the biology of this disease. From radiation therapy alone, the treatment has evolved to include multiple modalities. The outcome for localized disease has improved dramatically but more needs to be done for patients with metastatic or recurrent EWS. Targeted therapies may offer some hope for the latter group.
References
1. Diffuse endothelioma of bone. Ewing, JR. 1921, Proceedings of the New York Pathological Society, Vol. 21, pp. 17-24.
2. Ewing sarcoma: an eponym window to history. Cripe, TP. 2011, Sarcoma, Vol. 2011, pp. 1-4.
3. Ewing’s sarcoma. Karosas, AO. 2010, American Journal of Helath-System Pharmacy, Vol. 67, pp. 1599-1605.
4. Hawkins, DS, et al. Ewing sarcoma. [ed.] DG Poplack PA Pizzo. Principles and practice of pediatric oncology. 6th. Philadelphia : Lippincott, Williams and Wilkins, 2011, pp. 987-1014.
5. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. Esiashvili, N, Goodman, M and Marcus, Jr, RB. 2008, Journal of Pediatric Hematology/Oncology, Vol. 30, pp. 425-430.
6. Patterns of care and survival for patients aged under 40 years with bone sarcoma in Britain, 1980-1994. Stiller, CA, et al. 2006, British Journal of Cancer, Vol. 94, pp. 22-29.
7. Rarity of Ewing’s sarcoma among U.S. Negro children. Fraumeni, Jr, JF and Glass, AG. 1970, Lancet, Vol. 1, pp. 366-367.
8. Rarity of Ewing’s sarcoma in China. Li, FP, et al. 1980, Lancet, Vol. 1, p. 1255.
9. Prgnostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. Cotterill, SJ, et al. 2000, Journal of Clinical Oncology, Vol. 18, pp. 3108-3114.
10. Ushigome, S, Machinami, R and Sorensen, PH. Ewing sarcoma/primitive neuroectodermal tumor (PNET). [ed.] KK Unni, F Martens, eds CDM Fletcher. World Health Organization classification of tumours. Pathology and genetics. Tumours of soft tissues and bone. Lyon, France : IARC Press, 2002.
11. Identification of cancer stem cells in Ewing’s sarcoma. Suva, ML, et al. 2009, Cancer Research, Vol. 69, pp. 1776-1781.
12. The Ewing family of tumors-a subgroup of small-round-cell tumors defined by specific chimeric transcripts. Delattre, O, et al. 1994, The New England Journal of Medicine, Vol. 331, pp. 294-299.
13. Diagnosis and treatment of Ewing’s sarcoma. Iwamoto, Y. 2007, Japanese Journal of Clinical Oncology, Vol. 37, pp. 79-89.
14. Neuroectodermal differentiation in Ewing’s sarcoma family of tumors does not predict tumor behavior. Parham, DM, et al. 1999, Human Pathology, Vol. 30, pp. 911-918.
15. Cloning and characterization of Ewing’s sarcoma and peripheral neuroepithelioma t(11;22) translocation breakpoints. Zucman, J, et al. 1992, Genes Chromosomes and Cancer, Vol. 5, pp. 271-277.
16. Diagnositc value of the molecular genetic detection of the t(11;22) translocation in Ewing’s tumors. Dockhorn-Dworniczak, B, et al. 1994, Virchows Archives, Vol. 425, pp. 107-112.
17. Molecular analysis of Ewing’s sarcoma: another fusion gene, EWS-E1AF, available for diagnosis. Urano, F, et al. 1998, Japanese Journal of Cancer Research, Vol. 89, pp. 703-711.
18. Promiscuous partnerships in Ewing’s sarcoma. Sankar, S and Lessnick, SL. 2011, Cancer Genetics, Vol. 204, pp. 351-365.
19. Ploidy and karyotype complexity are powerful prognostic indicatiors in the Ewing’s sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children’s Cancer and Leukemia Group. Roberts, P, et al. 2008, Genes, Chromosomes and Cancer, Vol. 47, pp. 207-220.
20. Prognostic impact of deletions at 1p36 and numerical aberrations in Ewing tumors. Hattinger, CM, et al. 1999, Genes Chromosomes and Cancer, Vol. 24, pp. 243-254.
21. Diagnostic accuracy of 18F-FDG-PET and PET/CT in patients with Ewing sarcoma family tumors: a systematic review and a meta-analysis. Treglia, G, et al. 2012, Skeletal Radiology, Vol. 41, pp. 249-256.
22. An evaluation of [F-18]-fluorodeoxy-d-glucose positron emission tomography, bone scan, and bone marrow aspiration/biopsy as staging investigations in Ewing sarcoma. Newman, EN, et al. 2013, Pediatric Blood and Cancer, Vol. 60, pp. 1113-1117.
23. Futility versus utility of marrow assessment in initial Ewing sarcoma staging workup. Anderson, P. 2015, Pediatric Blood and Cancer, Vol. 62, pp. 1-2.
24. Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. Landenstein, R, et al. 2010, Journal of Clinical Oncology, Vol. 28, pp. 3284-3291.
25. https://www.clinicaltrials.gov/ct2/show/NCT01231906. ClinicalTrials.gov. [Online] [Cited: 5 June 2015.]
26. https://www.clinicaltrials.gov/ct2/show/NCT02306161. ClinicalTrials.gov. [Online] [Cited: 5 June 2015.]
27. Utility of bone marrow aspiration and biopsy in initial staging of Ewing sarcoma. kopp, LM, et al. 2015, Pediatric Blood and Cancer, Vol. 62, pp. 12-15.
28. Ewing’s sarcoma: a study of treatment methods. Jenkin, RD. 1966, Clinical Radiology, Vol. 17, pp. 97-106.
29. The curability of Ewing’s endothelioma of bone in children. Phillips, RF and Higinbotham, NL. 1967, Journal of Pediatrics, Vol. 70, pp. 391-397.
30. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. Grier, HE, et al. 2003, New England Journal of Medicine, Vol. 348, pp. 694-701.
31. Long-term survival in patients with Ewing’s sarcoma relapsing after completing therapy. Hayes, FA, et al. 1987, Medical and Pediatric Oncology, Vol. 15, pp. 254-256.
32. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Stahl, M, et al. 2011, Pediatric Blood and Cancer, Vol. 57, pp. 549-553.
33. Ewing sarcoma treatment. Jurgens, H and Dirksen, U. 2011, European Journal of Cancer, Vol. 47 Suppl 3, pp. S366-S367.
34. Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies-European Intergroup Cooperative Ewing Sarcoma Studies. Paulussen, M, et al. 1998, Annals of Oncology, Vol. 9, pp. 275-281.
35. Long-term results from the first UKCCSG Ewing’s tumor study (ET-1): United Kingdom Children’s Cancer Study Group (UKCCSG) and the Medical Research Council Bone Sarcoma Working Party. Craft, AW, et al. 1997, European Journal of Cancer, Vol. 33, pp. 1061-1069.
36. Long-term event-free survival after intensive chemotherapy for Ewing’s family of tumors in children and young adults. Kolb, EA, et al. 2003, Journal of Clinical Oncology, Vol. 21, pp. 3423-3430.
37. Analysis of prognostic factors in Ewing sarcoma family of tumors: review of St. Jude Children’s Research Hospital studies. Rodriguez-Galindo, C, et al. 2007, Cancer, Vol. 110, pp. 375-384.
38. Treatment of metastatic Ewing’s sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide-a Children’s Cancer Group and Pediatric Oncology Group study. Miser, JS, et al. 2004, Journal of Clinical Oncology, Vol. 22, pp. 2873-2876.
39. Ewing’s tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing’s Sarcoma Studies patients. Paulussen, M, et al. 1998, Journal of Clinical Oncology, Vol. 16, pp. 3044-3052.
40. Treatment of nonmetastatic Ewing’s sarcoma family tumors of the spine and sacrum: the experience from a single institution. Bacci, G, et al. 2006, European Spine Journal, Vol. 18, pp. 1091-1095.
41. Ewing tumors in infants. van den Berg, H, et al. 2008, Pediatric Blood and Cancer, Vol. 50, pp. 761-764.
42. Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: an analysis of 1631 cases from the SEER database, 1973-2005. Jawad, MU, et al. 2009, Cancer, Vol. 115, pp. 3526-3536.
43. Evaluation of prognostic factors in a tumor volume-adapted treatment strategy for localized Ewing sarcoma of bone: the CESS 86 experience. Ahrens, S, et al. 1999, Medical and Pediatric Oncology, Vol. 32, pp. 186-195.
44. Prognostic factors in non-metastatic Ewing’s sarcoma tumor of bone: an analysis of 579 patients treated at a single institution with adjuvant or neoadjuvant chemotherapy between 1972 and 1998. Bacci, G, et al. 2006, Acta Oncologica, Vol. 45, pp. 469-475.
45. [18F]Fluorodeoxyglucose positron emission tomography predicts outcome for Ewing sarcoma family of tumors. Hawkins, DS, et al. 2005, Journal of Clinical Oncology, Vol. 23, pp. 8828-8834.
46. Assessment of histological response of pediatric bone sarcomas using FDG PET in comparison to morphological volume measurement and standardized MRI parameters. Denecke, T, et al. 2010, European Journal of Nuclear Medicine and Molecular Imaging, Vol. 37, pp. 1842-1853.
47. Increased risk of systematic relapse associated with bone marrow micrometastasis and circulating tumor cells in localized Ewing tumor. Schleiermacher, G, et al. 2003, Journal of Clinical Oncology, Vol. 21, pp. 85-91.
48. Overexpression of p53 protein in primary Ewing’s sarcoma of bone: relationship to tumor stage, response and prognosis. Abudu, A, et al. 1999, British Journal of Cancer, Vol. 79, pp. 1185-1189.
49. Genetic imbalances revealed by comparative genomic hybridization in Ewing tumors. Ozaki, T, et al. 2001, Genes Chromosomes and Cancer, Vol. 2001, pp. 164-171.
50. Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. Scotlandi, K, et al. 2009, Journal of Clinical Oncology, Vol. 27, pp. 2209-2216.
51. Clinical features and outcomes in patients with secondary Ewing sarcoma. Applebaum, MA, et al. 2013, Pediatric Blood and Cancer, Vol. 60, pp. 611-615.
52. The histological response to chemotherapy as a predictor of the oncological outcome of operative treatment of Ewing sarcoma. Wunder, JS, et al. 1998, Journal of Bone and Joint Surgery, American volume, Vol. 80, pp. 1020-1033.
53. Chemotherapy response is an important predictor of local recurrence in Ewing sarcoma. Lin, PP, et al. 2007, Cancer, Vol. 109, pp. 603-611.
54. Allogeneic and autologous stem-cell transplantation in advanced Ewing tumors. An update after long-term follow-up from two centers of the European Intergroup study EICESS. . Burdach, S, et al. 2000, Annals of Oncology, Vol. 11, pp. 1451-1462.
55. Current treatment of Ewing’s sarcoma. Thacker, MM, et al. 2005, Expert Review of Anticancer Therapy, Vol. 5, pp. 319-331.
56. Ewing’s sarcoma-A critical analysis of 165 cases. Dahlin, DC, Coventry, MD and canlon, PW. 1961, Journal of bone and Joint Surgery, Vol. 43 (A), pp. 185-192.
57. Cyclophosphamide therapy in children with Ewin’g sarcoma. Sutow, WW and Sullivan, MP. 1962, Cancer Chemotherapy Reports, Vol. 23, pp. 55-60.
58. Cyclophosphamide in children with cancer. Pinkel, D. 1962, Cancer, Vol. 15, pp. 42-49.
59. Cyclophosphamide in the management of Ewing’s sarcoma. Samuels, ML and Howe, CD. 1967, Canver, Vol. 20, pp. 961-966.
60. Vincristine in children with malignant solid tumors. James Jr, DH and George, P. 1964, The Journal of Pediatrics, Vol. 64, pp. 534-541.
61. Daunomycin, an antitumor antibiotic, in the treatment of neoplastic disease. Clinical evaluation with special reference to childhood leukemia. Tan, C, et al. 1967, Cancer, Vol. 20, pp. 333-353.
62. Treatment of pulmonary metastatic disease with radiation therapy and adjuvant actinomycin D. Preliminary observations. Cupps, RE, Ahmann, DL and Soule, EH. 1969, Cancer, Vol. 24, pp. 719-723.
63. Treatment of clinically localized Ewing’s sarcoma with radiotherapy and combination chemotherapy. Hustu, HO, Pinkel, D and Pratt CB. 1972, Cancer, Vol. 30, pp. 1522-1527.
64. The response to initial chemotherapy as a prognostic factor in localized Ewing sarcoma. Oberlin, O, et al. 1985, European Journal of Clinical Oncology, Vol. 21, pp. 463-467.
65. Prognostic factors in localized Ewing tumors and peripheral neuroectodermal tumors: the third study of the French Society of Pediatric Oncology (EW88 study). Oberlin, O, et al. 2001, British Journal of Cancer, Vol. 85, pp. 1646-1654.
66. No benefit of ifosfamide in Ewing’s sarcoma: A nonrandomised study of the French Society of Pediatric Oncology. Oberlin, O, et al. 1992, Journal of Clinical Oncology, Vol. 10, pp. 1407-1412.
67. Ifosfamide-containing chemotherapy in Ewing’s sarcoma: the second United Kingdom Children’s Cancer Study Group and the Medical Research Council Ewing’s Tumor Study (ET-2). Craft, A, et al. 1998, Journal of Clinical Oncology, Vol. 16, pp. 3628-3633.
68. Ewing’s sarcoma treatment in Scandinavia 1984-1990: Ten-year results of the Scandinavian Sarcoma Group Protocol SSGIV. Nilbert, M, et al. 1998, Acta Oncologica, Vol. 37, pp. 375-378.
69. Five-year results in Ewing’s sarcoma. The Scandinavian Sarcoma Group experience with the SSG IX protocol. Elomaa, I, et al. 2000, European Journal of Cancer, Vol. 36, pp. 875-880.
70. Multidisciplinary treatment of primary Ewing’s sarcoma of bone. A 6-year experience of a European Cooperative Trial. Jurgens, H, et al. 1988, Cancer, Vol. 61, pp. 23-32.
71. Localized Ewing’s tumor of bone: final results of the Cooperative Ewing’s Sarcoma Study CESS 86. Paulussen, M, et al. 2001, Journal of Clinical Oncology, Vol. 19, pp. 1818-1829.
72. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term follow-up of the First Intergroup Study. Nesbit Jr, ME, et al. 1990, Journal of Clinical Oncology, Vol. 8, pp. 1664-1674.
73. Multimodality therapy for the management of localized Ewing’s sarcoma of pelvic and sacral bomes: a report from the Second Intergroup Study. Evans, RG, et al. 1991, Journal of Clinical Oncology, Vol. 9, pp. 1173-1180.
74. Multimodal therapy for the management of nonpelvic, localized Ewing’s sarcoma of bone: Intergroup Study IESS-II. Burgert Jr, EO, et al. 1990, Journal of Clinical Oncology, Vol. 8 , pp. 1514-1524.
75. Progress in the treatment of Ewing sarcoma: are the rumors of the demise of cytotoxic chemotherapy premature? Rosen, G. 2015, Klinische Padiatrie, Vol. 227, pp. 105-107.
76. Phase II study of VP-16-213 in childhood malignant disease: a Children’s Cancer Study Group report. Chard Jr, RL, et al. 1979, Cancer Treatment Reports, Vol. 63, pp. 1755-1759.
77. A phase II study of ifosfamide in children with recurrent solid tumors. Pinkerton, CR, Rogers, H and James, C. 1985, Cancer Chemotherapy and Pharmacology, Vol. 15, pp. 258-262.
78. Ifosfamide plus etoposide in newly diagnosed Ewing’s sarcoma of bone. Meyer, WH, et al. 1992, Journal of Clinical Oncology, Vol. 10, pp. 1737-1742.
79. Results of the EICESS-92 study: Two randomised trials of Ewing’s sarcoma treatment-Cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. Paulussen, M, et al. 2008, Journal of Clinical Oncology, Vol. 26, pp. 4385-4393.
80. Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-Ewing99-R1 trial. Le Deley, M, et al. [ed.] 2448. 2014, Journal of Clinical Oncology, Vol. 32, p. 2440.
81. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: A Children’s Oncology Group study. Granowetter, L, et al. 2009, Journal of Clinical Oncology, Vol. 27, p. 25362541.
82. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s Oncology Group. Womer, RB, et al. 2012, Journal of Clinical Oncology, Vol. 30, pp. 4148-4154.
83. Ifosfamide in combination chemotherapy for sarcomas and testicular carcinoma. Niederle, N, et al. 1985, Cancer Treatment Reviews, Vol. 10(Suppl 1), pp. 129-135.
84. Cyclophosphamide dose escalation in comparison with vincrstine and actinomycin-D (VAC) in gross residual sarcoma: A pilot study without hematopoietic growth factor support evaluating toxicity and response. Ruymann, FB, et al. 1995, Journal of Pediatric Hematology/Oncology, Vol. 17, pp. 331-337.
85. Long-term follow-up of ifosfamide renal toxicity in children treated for malignant mesenchymal tumors: An International Society of Pediatric Oncology report. Suarez, A, et al. 1991, Journal of Clinical Oncology, Vol. 9, pp. 2177-2182.
86. Second malignancies after Ewing’s sarcoma: radiation dose-dependency of secondary sarcomas. Kuttesch, JF Jr, et al. 1996, Journal of Clinical Oncology, Vol. 14, pp. 2818-2825.
87. Comparative evaluation of local control strategies in localized Ewing sarcoma of bone: a report from the Children’s Oncology Group . DuBois, SG, et al. 2015, Cancer, Vol. 121, pp. 467-475.
88. Current therapeutic approaches in metastatic and recurrent Ewing sarcoma. Huang , M and Lucas, K. 2011, Sarcoma.
89. The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Haeusler, J, et al. 2010, Cancer, Vol. 116, pp. 443-450.
90. High-risk Ewing’s sarcoma: end-intensification using autologous bone marrow transplantation. Marcus, RB, Jr, et al. 1988, International Journal of Radiation Oncology*Biology*Physics, Vol. 15, pp. 53-59.
91. Ewing’s sarcoma metastatic at diagnosis. Results and comparisons of two intergroup Ewing’s sarcoma studies. Cangir, A, et al. 1990, Cancer, Vol. 66, pp. 887-893.
92. Total-body irradiation and autologous bone marrow transplant in the treatment of high-risk Ewing’s sarcoma and rhabdomyosarcoma. Horowitz, ME, et al. 1993, Journal of Clinical Oncology, Vol. 11, pp. 1911-1918.
93. Therapy for metastatic ESFT: is it time to ask new questions? Snyder, KM and Mackall, CL. 2007, Pediatric Blood and Cancer, Vol. 49, pp. 115-116.
94. High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing’s sarcoma does not improve prognosis. Meyers, PA, et al. 2001, Journal of Clinical Oncology, Vol. 19, pp. 2812-2820.
95. How effective is dose-intensive/myeloablative therapy against Ewing’s sarcoma/primitive neuroectodermal tumor metastatic to bone or bone marrow?The Memorial Sloan-Kettering experience and a literature review. Kushner, B and Meyers, P. 2001, Journal of Clinical Oncology, Vol. 19, pp. 870-880.
96. Myeloablative radiochemotherapy and hematopoietic stem-cell rescue in poor-prognosis Ewing’s sarcoma. Burdach, S, et al. 1993, Journal of Clinical Oncology, Vol. 11, pp. 1482-1488.
97. Does consolidation with autologous stem cell transplantation improve the outcome of children with metastatic or relapsed Ewing sarcoma? Al-Faris, N, et al. 2007, Pediatric Blood and Cancer, Vol. 49, pp. 190-195.
98. High-dose chemotherapy and autologous peripheral blood stem cell transfusion for adult and adolescent patients with small round cell sarcomas. Yamada, K, et al. 2007, Bone Marrow Transplantation, Vol. 29, pp. 471-476.
99. High-dose busulfan/melphalan with autologous stem cell rescue in Ewing’s sarcoma. Atra, A, et al. 1997, Bone Marrow Transplantation, Vol. 20, pp. 843-846.
100. Impact of high-dose busulfan plus melphalan as consolidation in metastatic Ewing tumors: a study by the Scoiete Francaise des Cancers de l’Enfant. Oberlin, O, Rey, A and Desfachelles, AS. 2006, Journal of Clinical Oncology, Vol. 24, pp. 3997-4002.
101. Autologous stem cell trasplantation for high-risk Ewing’s sarcoma and other pediatric solid tumors. Fraser, CJ, et al. 2006, Bone Marrow Transplantation, Vol. 37, pp. 175-181.
102. High-dose therapy with hematopoietic stem cell rescue in patients with poor prognosis Ewing family tumors. Rosenthal, J, et al. 2008, Bone Marrow Transplantation, Vol. 42, pp. 311-318.
103. Risk adapted chemotherapy for localised Ewing’s sarcoma of bone: the French EW93 study. Gasper, N, et al. 2012, European Journal of Cancer, Vol. 48, pp. 1376-1385.
104. Consolidation of first-line therapy with busulfan and melphala, and autologous stem cell rescue in children with Ewing’s sarcoma. Drabko, K, et al. 2012, Bone Marrow Transplantation, Vol. 47′, pp. 1530-1534.
105. High-dose therapy for patients with primary multifocal and early relapsed Ewing’s tumors: results of two consecutive regimens assessing the role of total-body irradiation. Burdach, S, et al. 2003, Journal of Clinical Oncology, Vol. 21, pp. 3072-3078.
106. Gene-marking to trace origin of relapse after autologous bone-marrow transplantation. Brenner, MK, et al. 1993, Lancet, Vol. 341, pp. 85-86.
107. Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: a report from the Children’s Oncology Group. Leavey, PJ, et al. 2008, Pediatric Blood and Cancer, Vol. 51, pp. 334-338.
108. Late recurrence in pediatric cancer: a report from the Childhood Cancer Survivor Study. Wasilewski-Masker, K, et al. 2009, journal of National Cancer Institute, Vol. 101, pp. 1709-1720.
109. Ewing’s sarcoma family of tumors: current management. Bernstein, M, et al. 2006, Oncologist, Vol. 11, pp. 503-519.
110. Ewing’s sarcoma: standard and experimental treatment options. Subbiah, V, et al. 2009, Current Treatment Options in Oncology, Vol. 10, pp. 126-140.
111. Response to high dose ifosfamide in patients with advanced/recurrent Ewing’s tumors. Ferrari, S, et al. 2009, Pediatric Blood and Cancer, Vol. 52, pp. 581-584.
112. Survival after recurrence of Ewing tumors: The St. Jude Children’s Research Hospital experience, 1979-1999. Rodriguez-Galindo, C, et al. 2002, Cancer, Vol. 94, pp. 561-560.
113. Ifosfamide, carboplatin and etoposide (ICE) reinduction chemotherapy in a large cohort of children and adolescents with recurrent/refractory sarcoma: the Children’s Cancer Group (CCG) experience. Van Winkle, P, et al. 2005, Pediatric Blood and Cancer, Vol. 44, pp. 338-347.
114. Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. Saylors, RLIII, et al. 2001, Journal of Clinical Oncology, Vol. 19, pp. 3463-3469.
115. Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Hunold, A, et al. 2006, Pediatric Blood and Cancer, Vol. 47, pp. 795-800.
116. Cyclophosphamide and topotecan as first-line salvage therapy in patients with relapsed Ewing sarcoma at a single institution . Farhat, R, et al. 2013, Journal of Pediatric Hematology/Oncology, Vol. 35, pp. 356-360.
117. Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Wagner, LM, et al. 2007, Pediatric Blood and Cancer, Vol. 48, pp. 132-139.
118. Irinotecan and temozolomide for Ewing sarcoma. Casey, DA, et al. 2009, Pediatric Blood and Cancer, Vol. 2009, pp. 1029-1034.
119. Vincristine, irinotecan, and temozolomide in patients with relapsed and refractory Ewing sarcoma. Raciborska, A, et al. 2013, Pediatric Blood and Cancer, Vol. 60, pp. 1621-1625.
120. Irinotecan and Temozolomide treatment for relapsed Ewing sarcoma: a single center experience and review of the literature. Kurucu, N, Sari, N and Ilhan, IE. 2015, Pediatric Hematology and Oncology, Vol. 32, pp. 50-59.
121. Treatment of relapsed/refractory pediatric sarcomas with gemcitabine and docetaxel. Mora, J, et al. 2009, Journal of Pediatric Hematology/Oncology, Vol. 31, pp. 723-729.
122. Phase II study of sequential gemcitabine followed by docetaxel for recurrent Ewing sarcoma, osteosarcoma, or unresectable or locally recurrent chondrosarcoma: results of Sarcoma Alliance for Research Through Collaboration tudy 003. Fox, E, et al. 2012, Oncologist, Vol. 17, p. 321.
123. Survival after recurrence of Ewing’s sarcoma family of tumors. Barker, LM, et al. 2005, Journal of Clinical Oncology, Vol. 23, pp. 4354-4362.
124. The value of high-dose chemotherapy in patients with first relapsed Ewing sarcoma. Rasper, M, et al. 2014, Pediatric Blood and Cancer, Vol. 61, pp. 1382-1386.
125. High dose chemotherapy with bone marrow or peripheral stem cell rescue is an effective treatment option for patients with relapsed or progressive Ewing’s sarcoma family of tumors. McTiernan, A, et al. 2006, Annals of Oncology, Vol. 17, pp. 1301-1305.
126. Myeloablative therapy with autologous stem cell rescue for Ewing sarcoma. Gardner, SL, et al. 2008, Bone Marrow Transplantation, Vol. 41, pp. 867-872.
127. Role of tyrosine kinase inhibitors in cancer therapy. Arora, A and Scholar, EM. 2005, Journal of Pharmacology and Experimental Therapeutics, Vol. 315, pp. 971-979.
128. The role of IGF1R in pediatric malignancies. KIm, SY, et al. 2009, Oncologist, Vol. 14, pp. 83-91.
129. IGF-1R taargeted treatment of sarcoma. Toretsky, JA and Gorlick, R. 2010, The Lancet Oncology, Vol. 11, pp. 1015-106.
130. Targeted therapy for Ewing’s sarcoma. Subbiah, V and Anderson, P. 20011, Sarcoma.
131. R1507,a monoclonal antibody to the insulin-like grwoth factor 1 receptor,in patients with recurrent or refractory Ewing sarcoma family of tumors:results of a phase II Sarcoma Alliance for Research through Collaboration study. Pappo, AS, et al. 2011, Journal of Clinical Oncology, Vol. 29, pp. 4541-4547.
132. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. Jurgens, H, et al. 2011, Journal of Clinical Oncology, Vol. 29, pp. 4534-4540.
133. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. Tap, WD, et al. 2012, Journal of clinical oncology, Vol. 30, pp. 1849-1856.
134. Phase I/II trial and pharmakokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children’s Oncology Group. Melampati, S, et al. 2012, Journal of Clinical Oncology, Vol. 30, pp. 256-262.
135. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing’s sarcoma family tumors. Naing, A, et al. 2012, Clinical Cancer Research, Vol. 18, pp. 2625-2631.
136. Activity of SCH-717454 in subjects with relapsed osteosarcoma or Ewing’s sarcoma (study P04720). Anderson, P, et al. London, UK : s.n., 2007. Proceedings of the 14th Annual Meeting of the Connective Tissue Oncology Society (CTOS ’07). Abstract #35094.
137. Targeting the insulin-like growth factor 1 receptor in Ewing’s sarcoma: reality and expectations. Olmos, D, et al. 2011, Sarcoma.
138. GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Sabbatini, P, et al. 2009, Molecular Cancer Therapeutics, Vol. 8, pp. 2811-2820.
139. Antitumor activity of GSK1904529A, a small-molecule inhibitor of the insulin-like growth factor-I receptor tyrosine kinase. Sabbatini, P, et al. 2009, Clinical Cancer Research, Vol. 15, pp. 3058-3067.
140. BMS-754807, a small molecule inhibotor of insulin-like growth factor-1R/IR. Carboni, JM, et al. 20009, Molecular Cancer Therapeutics, Vol. 8, pp. 3341-3349.
141. Biological rationale and current clinical experience with anti-insulin-like growth factor 1 receptor monoclonal antibodies in treating sarcoma: twenty years from the bench to the bedside. Olmos, D, et al. 2010, Cancer Journal, Vol. 16, pp. 183-194.
142. A phase II study of imatinib mesylate in children with refractory or relapsed solid tumors: a Children’s Oncology Group study. Bond, M, et al. 2008, Pediatric Blood and Cancer, Vol. 50, pp. 254-258.
143. Phase II multicentre trial of imatinib in 10 histologic subtypes of sarcoma using a Bayesian Hierarchical statistical model. Chugh, R, et al. 2009, Journal of Clinical Oncology, Vol. 27, pp. 3148-3153.
144. Phase II clinical trial of imatinib mesylate in therapy of KIT and/or PDGFRa-expressing Ewing sarcoma family of tumors and desmoplastic small round cell tumors. Chao, J, et al. 2010, Anticancer Research, Vol. 30, pp. 547-552.
145. Results of a Sarcoma Alliance for Research through Collaboration (SARC) phase II trial of dasatinib in previously treated, high-grade, advanced sarcoma. Schuetze, S, et al. 15s, 2010, Journal of Clinical Oncology, Vol. 28, p. abstract 10009.
146. Novel bone cancer drugs: investigational agents and control paradigms for primary bone sarcomas (Ewing’s sarcoma and osteosarcoma). Anderson, P, et al. 2008, Expert Opinion on Investigational Drugs, Vol. 17, pp. 1703-1715.
147. Suppression of Ewing’s sarcoma tumor growth, tumor vessel formation, and vasculogenesis following anti-vascular endothelial growth factor receptor-2 therapy. Zhou, Z, et al. 2007, Clinical Cancer Research, Vol. 13, pp. 4867-4873.
148. Phase I trial and pharmacokinetic study of bevacizumab in pediatric patients with refractory solid tumors: a Children’s Oncology Group study. Bender, JLG, et al. 2008, Journal of Clinical Oncology, Vol. 26, pp. 399-405.
149. Feasibility of bevacizumab (NSC 704865, BB-IND# 7921) combined with vincristine, topotecan, and cyclophosphamide in patients with first recurrent Ewing sarcoma (EWS): A Children’s Oncology Group (COG) study. Leavey, P, et al. 15s, 2010, Journal of Clinical Oncology, Vol. 28, p. abstract#9552.
150. A pilot study of low-dose anti-angiogenic chemotherapy in combination with standard multiagent chemotherapy for patients with newly diagnosed metastatic Ewing sarcoma family of tumors: a Children’s Oncology Group (COG) phase II study NCT00061893. Felgenhauer, JL, et al. 2013, Pediatric Blood and Cancer, Vol. 60, pp. 409-414.
151. Antitumor effects of histone deacetylase inhibitor on Ewing’s family of tumors. Sakimura, R, et al. 2005, International Journal of Cancer, Vol. 116, pp. 784-792.
152. Phase 1 Trial of Temsirolimus in Combination with Irinotecan and Temozolomide in Children, Adolescents and Young Adults with Relapsed or Refractory Solid Tumors: A Children’s Oncology Group Study. Bagatell, R, et al. 2014, Pediatric Blood and Cancer, Vol. 61, pp. 833-839.
153. Phase II study of cixutumumab in combination with temsirolimus in pediatric patients and young adults with recurrent or refractory sarcoma: a report from the Children’s Oncology Group. Wagner, LM, et al. 2015, Pediatric Blood and Cancer, Vol. 62, p. 440.
154. Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. Demetri, GD, et al. 2013, Journal of Clinical Oncology, Vol. 31, pp. 2484-2492.
155. Initial Testing of the Aurora Kinase A Inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Maris, JM, et al. 2010, Pediatric Blood and Cancer, Vol. 55, pp. 26-34.
156. https://clinicaltrials.gov/ct2/show/NCT01154816. ClinicalTrials.gov. [Online] [Cited: 5 May 2015.]
157. Arsenic trioxide inhibits Ewing’s sarcoma cell invasiveness by targeting p38(MAPK) and c-Jun N-terminal kinase. Zhang, S, et al. 2012, Anticancer Drugs, Vol. 23, pp. 108-118.
158. Mechanism of action of bisphosphonates on tumor cells and prospects for use in the treatment of malignant osteolysis. Clezardin, P, Gligorov, J and Delmas, P. 2000, Joint Bone Spine, Vol. 67, pp. 22-29.
159. Zoledronic acid inhibits primary bone tumor growth in Ewing sarcoma. Zhou, Z, et al. 2005, Cancer, Vol. 104, pp. 1713-1720.
160. Histone deacetylase inhibitors enhance expression of NKG2D ligands in Ewing sarcoma and sensitize for natural killer cell-mediated cytolysis. Berghuis, D, et al. 2012, Clinical Sarcoma Research, Vol. 2.
161. Killing the killer: natural killer cells to treat Ewing’s sarcoma. Ahn, YO, Weigel, B and Verneris, MR. 2010, Clinical Cancer Research, Vol. 16, pp. 3819-3821.
162. Ewing’s sarcoma: overcoming the therapeutic plateau. Subbiah, V and Kurzrock, R. 2012, Discovery Medicine, Vol. 13, pp. 405-415.
163. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Erkizan, HV, et al. 2009, Nature Medicine, Vol. 15, pp. 750-756.
164. Single enantiomer of YK-4-279 demonstrates specificity in targeting the oncogene EWS-FLI1. Barber-Rotenberg, JS, et al. 2012, Oncotarget, Vol. 3, pp. 172-182.
165. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. Grohar, PJ, et al. 2011, Journal of the National Cancer Institute, Vol. 103, pp. 962-978.
166. Small-molecule screen identifies modulators of EWS/FLI1 target gene expression and cell survival in Ewing’s sarcoma. Boro, A, et al. 2012, International Journal of Cancer, Vol. 131, pp. 2153-2164.
| How to Cite this article: Valvi S & Kellie SJ. Ewing Sarcoma: Focus on Medical Management. Journal of Bone and Soft Tissue Tumors May-Aug 2015;1(1):8-17. |
 - Dr. Santosh Valvi
|
 - Dr. Stewart J Kellie
|
Like this:
Like Loading...