Volume 2 | Issue 1 | Jan-Apr 2016 | Page 39-43 | Ashish Gulia, Pankaj Kumar Panda.
Author: Ashish Gulia [1] , Pankaj Kumar Panda [2]
[1]Orthopaedic Oncologist, Tata Memorial Hospital, Mumbai.
[2]Post-graduate Student, Clinical Research, Tata Memorial Hospital, Mumbai
Address of Correspondence
Dr Ashish Gulia
Associate Professor, Orthopedic Oncology, Dept. of Surgical Oncology, Tata Memorial Hospital,
Mumbai – 400012, India.
Email: aashishgulia@gmail.com
website: animaljam2.net
Abstract
Background: Fibrous dysplasia (FD) belongs to a group of non-hereditary benign pathologies in which immature bone and fibrous stroma replaces normal medullary bone. The gene for FD is located on band 20q13, an area that codes for the α subunit on G-protein receptors. It is most commonly diagnosed in the first three decades of life. Amongst the 4 major clinical forms of FD (namely monostotic, polyostotic, McCune-Albright syndrome, Mazabraud’s syndrome) the monostotic form predominates (70-80%) in comparison to the polyostotic form. “Ground Glass appearance is a characteristic appearance on plain radiograph. Histopathologically fibrous component is relatively avascular, composed of cytologically bland spindle cells with few trabecular structures. The management ranges from watchful observation to surgical intervention.
Key-words: Fibrous dysplasia, polyostotic, monostotic, bone grafting, bisphosphonates.
Introduction
Fibrous dysplasias (FD) are a group of non-hereditary benign pathologies in which immature bone and fibrous stroma replace normal medullary bone as a result of abnormal differentiation of osteoblasts characterized by solitary (monostotic) or multi focal medullary (polyostotic) fibro osseous lesions. They contain mutated fibroblast cells and osteoblasts of varying functionality, which produce abnormally immature woven bone [1]. They are accounted for 2.5% of all the bone tumors and 5–7% of all benign bone tumors [2]. The detailed description as a benign developmental disorder of the bone was given by Lichtenstein and Jaffe in 1942 and thus it is referred as Lichtenstein-Jaffe disease [3]. The dormancy of FD increases from childhood to adulthood, however there is a life time risk of malignant transformation that of around 1–4% [4].
Etiopathogenesis
Cessation of bone maturation process at the stage of woven bone formation leading to inability to produce mature lamellar bone accounts for development of fibrous dysplasia. There have been many theories which have tried to explain the genesis of fibrous dysplasia. Excessive production of interleukin-6 production at local site has been related to increased resorption of bone by increasing the numbers of osteoclasts in these lesions. Genetic theory postulates that somatic mutation early in embryonic life causes a gene mosaicism. The earlier the mutation occurs, the more widespread the effects will be. The gene is located on band 20q13, an area that codes for the α subunit on G-protein receptors Mutations in the gene (GNAS I) result in a cascade which may lead to alteration in cellular differentiation and osteoblastic proliferation [5, 6]. According to hormonal theory, osteoblasts in fibrous dysplastic lesions have an elevated number of hormone receptors and thus have altered responses of bone formation. Hormonal alteration occurring during life, like in pregnancy usually sees exuberated growth of these lesions, thus explaining its hormonal genesis. Another theory explains that cAMP also activates Fos, which inhibits osteoblastic specific genes as well as stimulating cytokines that promote bone resorption by osteoclasts. Hypophosphatemia/phosphaturia, sometimes found in FD, is caused by excess secretion of a phosphatonin fibroblast growth factor [7].
Clinical Presentation and evaluation
FD is most commonly observed between 3 to 15 years age group, and majority of the cases are diagnosed in the first three decades of life. Polyostotic lesions usually present earlier as they are associated with a more severe form of the disease. Males and females are equally affected [9, 10]. Majority of patients with the polyostotic form of FD are symptomatic before the age of 10 years [7]. The monostotic form predominates (70-80%) in comparison to the polyostotic form [8]. Site affection may vary with the form of the disease. According to the sites of involvement for the polyostotic form femur, tibia are most commonly involved followed by skull and facial bones, pelvis, rib, humerus, radius and ulna, lumbar spine, clavicle, and cervical spine. The lesions may be unilateral or, less commonly, bilateral. Symptoms are related to the severity of the disease. Monostotic lesions may be asymptomatic and found incidentally. Polyostotic lesions present early with more symptoms in 60% of the patients. Pain along with swelling and deformity are the most common presenting complaints which is due to structural weakness and micro fractures in the affected bone. Deformity occurs due to abnormal bone growth or micro fractures and subsequent remodeling. Deformities of the lower limb especially the proximal femur can cause an antalgic gait and have a high risk of developing leg length discrepancies and pathological fractures. Common deformities are varus deformity of the proximal femur also known as the “shepherd’s crook deformity”, tibial bowing, bossing of the skull, prominent jaw, rib and chest wall masses. Symptoms may get exacerbated during pregnancy [7, 11].

Four types of fibrous dysplasias have been reported so far based on clinical conditions [11].
Monostotic FD: This represents around 70-80% of all FDs. This is the only form where craniofacial bones are affected. It occurs mostly in the age group of 20-30 years. Non-osteogenetic fibroma, aneurysmal bone cyst, giant cell tumor of bone, adamantinoma, eosionophilic granuloma and plasma cell myeloma should be considered in the differential diagnosis of monostotic FD [8, 12]
Polysototic FD: It accounts for 20-25% of all FDs. More than one of the bones in the skeletal and craniofacial system is affected and it occurs in the first decade of life. Hyperparathyroidism, polyostotic Paget’s disease, neurofibromatosis and cherubism should be considered in the differential diagnosis of polyostotic FD [8, 12].
McCune-Albright syndrome: It is a triad of polyostotic fibrous dysplasia, cutaneous café-au-lait spots and endocrine dysfunction. The syndrome was named after 2 physicians, Donovan McCune and Fuller Albright, who separately described the triad in 1937. It accounts for about 3% of all fibrous dyplasias and 35% to 50% of cases of polyostotic fibrous dysplasias. Females are affected more than males. Patient suffering from this syndrome will have hyperpigmented skin lesions with irregular “coast of Maine” borders with ipsilateral bony ground glass lesions. Endocrinopathies include hyperprolactinemia, gonadotropin-independent precocious puberty, growth hormone excess, hyperthyroidism, FGF23-mediated renal phosphate wasting and Cushing’s syndrome [13].
Mazabraud’s syndrome: Patients suffering from this syndrome present with soft tissue myxomas with polyostotic fibrous dysplasia. The myxomas generally develop later than FD adjacent to the affected bones. These are more commonly seen in extremities alongside long bones. [14].
Cherubism: It is an autosomal dominant disorder which is characterized by symmetric involvement of both the mandible and maxilla and manifests during the second decade of life. These lesions generally become static at skeletal maturity [15].
Radiological evaluation
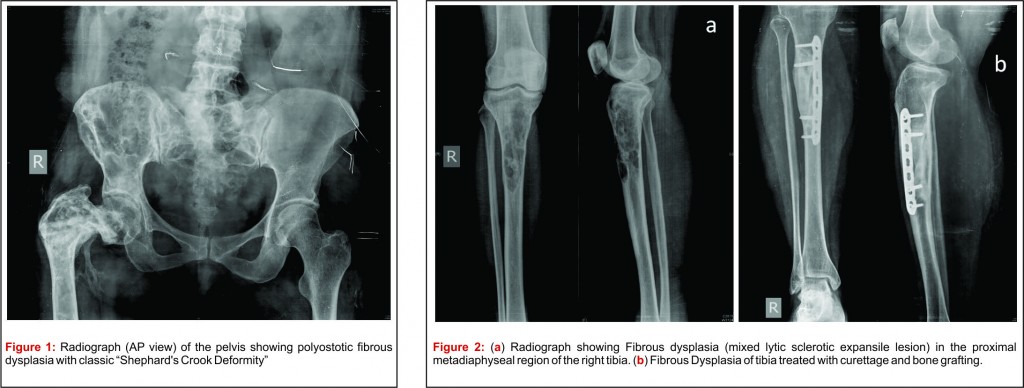
Plain radiograph: Plain radiograph is the gold standard to evaluate a fibrous dysplasia lesion. It typically shows a well defined medullary lesion, which is usually mildly expansile and is centered in either metaphysis or diaphysis with or without endosteal scalloping with a varying degree of translucency. The medullary canal is replaced with fibrous tissue formed of delicate woven bone spicules that give the tissue its “ground glass” appearance. There may be endosteal scalloping of the inner cortex, but the periosteal surface is smooth and nonreactive. [16]. The deformities (shephard’s crook curvature of femur and coxa vara deformity of the knee) may vary in severity (Figure 1: Radiograph (AP view) of the pelvis showing polyostotic fibrous dysplasia with classic “Shephard’s Crook Deformity”). Radiological characteristics of the lesions differ with respect to the bone and fibrous matrix ratio and are usually seen as three different patterns. Firstly the pagetoid pattern where the rate of the bone–fibrous matrix is equal, secondly sclerotic pattern in which the bone structure is in the foreground and thirdly the radiolucent pattern where the fibrous matrix is in the foreground [17].
Computed tomography: A CT scan demonstrates the extent of the lesion. The appearance may vary according to the amount of calcification and ossification in the lesion. CT Imaging may be more useful in evaluation of craniofacial FDs. [11].
Bone scintigraphy: It is helpful in detecting the extent of disease and distribution particularly of active lesions in adolescent period [18]. It may also be helpful in detecting stress fractures.
Magnetic resonance Imaging (MRI): MRI is helpful in assessing the exact location, extent, shape and content of FD lesion. It characteristically demonstrates a low-intensity signal on T1-weighted images due to its fibrous content. T2-weighted images demonstrate moderately intense signals which are darker than signal of malignant tissue, fat or fluid. MRI is also helpful in detecting malignant transformation of a FD lesion, which may be evident with features like cortical bone erosion, destruction and soft tissue masses [19].
Histopathological evaluation
The lesions show an expanded bone with well-circumscribed, tan grey mass that is dense and variably fibrous with a gritty consistency due to the presence of bone trabeculae. It may show cystic areas in older lesions with some yellow-tinged fluid. A glassier, blue-tinged appearance may be found in cases with chondroid metaplasia [7].
Microscopic appearance: Microscopic evaluation shows varying proportions of fibrous and osseous tissue. The fibrous component is avascular and composed of cytologically bland spindle cells demonstrating low mitotic rate without atypia. Trabecular structures show an abnormal arrangement resembling Chinese letters which is composed of woven bone. Secondary myxoid and aneurysmal bone cyst like changes can be seen. Occasional nodules of benign hyaline cartilage may also be seen. Osteoblastic cells create fibrous tissue instead of a normal bone tissue in the bone medulla [20, 21]
Differential Diagnosis
The differential diagnosis of FD varies based on location, extent of lesion and age of the patient. These mainly include simple bone cyst, enchondroma, eosinophilic granuloma, brown tumor of hyperparathyroidism, giant cell tumor, neurofibromatosis, osteoblastoma, hemangioma of bone. osteofibrous dysplasia, fracture callus, non-ossifying fibroma, and low-grade Osteosarcoma can be included in the differential diagnosis based on histopathological findings. Low-grade chondrosarcoma may be part of the differential diagnosis if there is a prominent chondroid component. Osteofibrous dysplasia and fracture callus can be differentiated by the history and location of the lesion, and they typically have prominent osteoblastic rimming around the bone trabeculae[7].
Treatment
Management of FD should focus on reducing pain, optimizing function, and managing endocrinopathies, if they exist. The choice of treatment is usually guided by, site & extent of lesion, growth of lesion, related symptoms and age of the patient; it can vary from a watchful observation to surgical intervention. Monostotic asymptomatic lesions can be observed and followed up with serial radiographs to look for progression of the lesion. Large symptomatic lesions especially in the lower limb require active management, which may include non surgical (medical) or surgical management. Surgical interventions should be dealt with caution as complete resections are not possible mostly and incomplete resections have high chances of recurrence [22]. Medical management with bisphosphonates is helpful in most sympotomatic monostotic lesions without fractures or deformity. Third generation bisphosphonates (zolendronic acid) have shown remarkable success rate in relieving bone pain and healing of lesions. This is radiologically evident by improved cortical thickness, ossification of the lesion and improved function. These have also shown to reduce the rate of complications like pathological fracture. Intravenous 4 mg zolendronic acid along with vitamin D and oral calcium supplements is the choice of treatment [23]. Denosumab also appears to be effective in reducing bone turnover in adult patients with active FD. However, caution should be exercised, and patients should be monitored carefully as significant fluctuations in biochemical and hormonal indices can occur [24] and hence Denosumab is not recommended for regular use. Surgical intervention is indicated in cases of failure of nonsurgical therapy, large painful lesions, progressive deformity, non-union or malignant transformation. Curettage and bone grafting is the main cornerstone of surgical management. Cortical strut allograft should be used whenever possible as cancellous grafts may get resorbed due to natural disease process. Appropriate internal fixation should be used judiciously. Deformities especially in lower limb require surgical correction. Valgus osteotomy and medial displacement osteotomy are used to correct these deformities. (Figure 2: (a) Radiograph showing Fibrous dysplasia (mixed lytic sclerotic expansile lesion) in the proximal metadiaphyseal region of the right tibia. (b) Post treatment radiograph showing curettage and bone grafting and interfixation) [24].
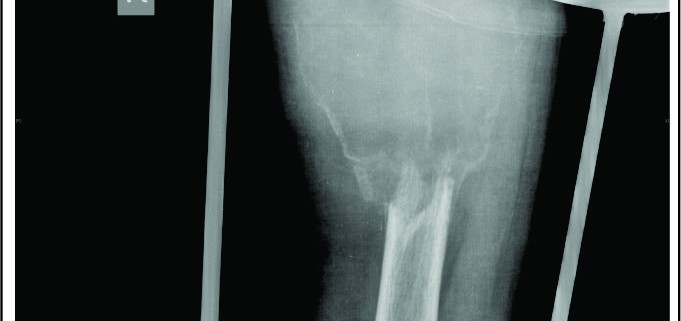
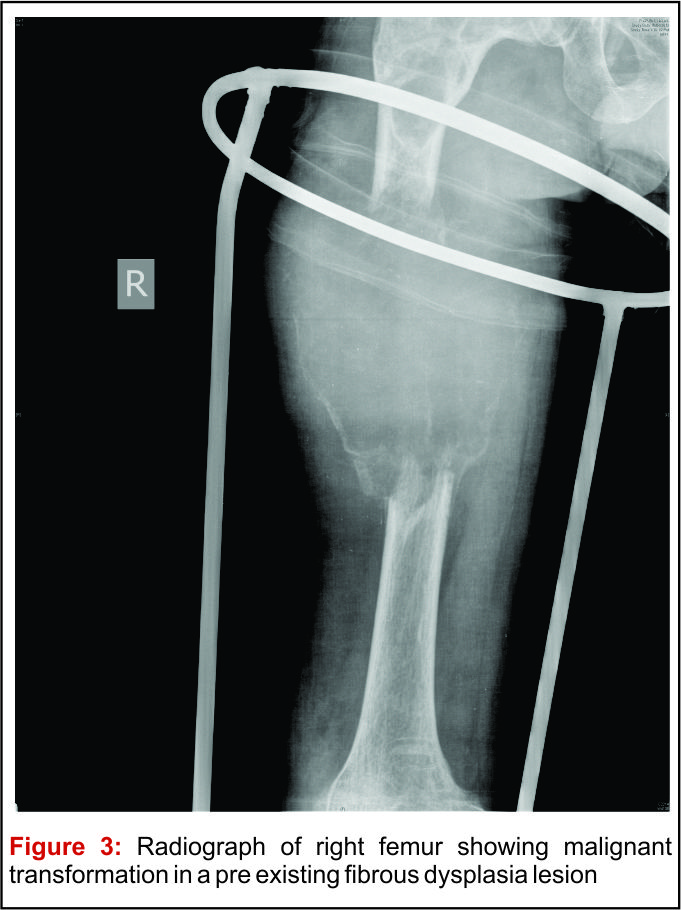
Prognosis: Generally the prognosis of FD patient is excellent in the absence of malignant transformation. Monostotic FDs have better prognosis than polyostotic or syndromal FDs. Medical management is also more successful in monostotic lesions. Femoral lesions, younger age group patients, polyostotic disease and surgical interventions without internal fixation are the negative prognostic factors for surgical management. Malignant transformation is rare (Figure 3: Radiograph of right femur showing malignant transformation in a pre existing fibrous dysplasia lesion) [25].
Complications: Main complications related to FD are uncontrolled pain, deformities, pathological fractures, limb length discrepancy and malignant transformation. Malignant transformation commonly occurs to osteosarcoma or fibrosarcoma. It generally presents with increase intensity of pain with associated progressively increasing mass with or without pathological fracture. It is more common in polyostotic disease with Endocrinopathies. The rates of malignant transformation have been estimated to be about 0.5% with monostotic FD and about 4% with McCune-Albright syndrome [26]. Prognosis is usually poor. These are managed with multimodality treatment including surgery and chemotherapy [27]
Conclusion
Fibrous Dysplasia even though has good prognosis, there is a wide range of severity in patients all over. While some are minimally affected, some present with numerous fractures and significant deformities. With the advent of advanced imaging modalities and molecular pathology, a better understanding of the pathogenesis of FD has been possible. Non-surgical treatment regimens are increasingly being followed owing to their better compliance and overall improvement in patients’ quality of life by minimizing pain.
References
1. Araghi HM, Haery C. Fibro-osseous lesions of craniofacial bones. The role of imaging. Radiol Clin North Am 1993 Jan;31(1):121–34.
2. Gupta A, Mehta VS, Sarkar C. Large cystic fibrous dysplasia of the temporal bone: case report and review of literature. J Clin Neurosci 2003;10(3):364–7.
3. Lichtenstein L. Polyostic fibrous dysplasia. Arch Surg 1938;36:874.
4. StantonRP. Surgery for fibrous dysplasia. J Bone Miner Res 2006; 21(Suppl 2): P105–P109.
5. Diaz A, Danon M, Crawford J. McCune-Albright syndrome and disorders due to activating mutations of GNAS1. J Pediatr Endocrinol Metab. 2007; 20(8): 853–880.
6. Lietman SA, Schwindinger WF, Levine MA. Genetic and molecular aspects of McCune-Albright syndrome. Pediatr Endocrinol Rev. 2007; 4(suppl 4):380–385.
7. Riddle ND , Bui MM. Fibrous Dysplasia. Arch Pathol Lab Med. 2013;137:134–138
8. Grabias SL, Campbell CJ. Fibrous dysplasia. Orthop Clin North Am 1997; 8:771–83.
9. Sharma RS, Mahapatra AK, Pawar SJ, et al. Symptomatic cranial fibrous dysplasia: clinico-radiological analysis in a series of 8 operative cases with follow-up results. J Clin Neurosci 2002; 9(4):381–90.
10. Rajendran R, Sivapathasundharam R. Shafer’s textbook of oral pathology 5th edition. New Delhi, India: Elsevier, a division of Reed Elsevier India Private Limited; 2006. p. 971–9.
11. DiCaprio MR, Enneking WF. Fibrous dysplasia: pathophysiology, evaluation, and treatment. J Bone Joint Surg Am. 2005; 87(8):1848–1864.
12. Resnick D. Diagnosis of bone and joint disorders. 4th ed. Philadelphia, PA: Saunders; 2002. 4285–4840.
13. Campanacci M. Bone and soft tissue tumors: clinical features, imaging, pathology and treatment. 2nd ed. Wien, Austria: Springer; 1999. p. 435–60.
14. Iwasko N, Steinbach LS, Disler D, et al. Imaging findings in Mazabraud’s syndrome: seven new cases. Skeletal Radiol 2002; 31:81–7.
15. Ruggieri P, Sim FH, Bond JR, et al. Malignancies in fibrous dysplasia. Cancer 1994;73:1411–24.
16. Nager GT, Kennedy DW, Kopstein E. Fibrous dysplasia: a review of the disease and its manifestations in the temporal bone. Ann Otol Rhinol Laryngol 1982(Suppl 92):1–52.
17. Fitzpatrick KA, Taljanovic MS, Speer DP, Graham AR, Jacobson JA, Barnes GR, Hunter TB. Imaging findings of fibrous dysplasia with histopathologic and intraoperative correlation. AJR Am J Roentgenol 2004; 182:1389–98.
18. Zhibin Y, Quanyong L, Libo C, Jun Z, Hankui L, Jifang Z, Ruisen Z. The role of radionuclide bone scintigraphy in fibrous dysplasia of bone. Clin Nucl Med 2004; 29:177–80.
19. Yavuzer R, Khilnani R, Jackson IT, Audet B. A case of atypical McCune-Albright syndrome requiring optic nerve decompression. Ann Plast Surg 1999; 43(4): 430-5.
20. Parekh SG, Donthineni-Rao R, Ricchetti E, Lackman RD. Fibrous dysplasia. J Am Acad Orthop Surg 2004; 12(5):305–13.
21. Sargin H, Gozu H, Bircan R, Sargin M, et al. A case of McCune-Albright syndrome associated with Gs alpha mutation in the bone tissue. Endocr J 2006; 53(1):35–44.
22. Amit M, Collins MT, FitzGibbon EJ, Butman JA, Fliss DM, Gil Z 2011 Surgery versus watchful waiting in patients with craniofacial fibrous dysplasia–a meta-analysis. PLoS One. 2011;6(9):e25179
23. Wu Di, Ma Jie , Bao Suqing , Guan Haixia. Continuous effect with long-term safety in zoledronic acid therapy for polyostotic fibrous dysplasia with severe bone destruction. Rheumatol Int. 2015 Apr;35(4):767-72
24. Ganda K, Seibel MJ. Rapid biochemical response to denosumab in fibrous dysplasia of bone: report of two cases. Osteoporos Int. 2014 Feb;25(2):777-82
25. Yabut SM Jr, Kenan S, Sissons HA, Lewis MM. Malignant transformation of fibrous dysplasia. A case report and review of the literature. Clin Orthop Relat Res. 1988; 228:281-9.
26. Jhala DN, Eltoum I, Carroll AJ, et al. Osteosarcoma in a patient with McCune-Albright syndrome and Mazabraud’s syndrome: a case report emphasizing the cytological and cytogenetic findings. Hum Pathol. 2003; 34(12):1354–1357.
27. Bielack SS, Kempf-Bielack B, Heise U, Schwenzer D, Winkler K; Cooperative German-Austrian-Swiss Osteosarcoma Study Group. Combined modality treatment for osteosarcoma occurring as a second malignant disease. J Clin Oncol 1999; 17(4):1164.
28. Eversole R, Su L, ElMofty S. Benign fibro-osseous lesions of the craniofacial complex. A review. Head Neck Pathol. 2008 Sep;2(3):177-202
| How to Cite this article: Gulia A, Panda PK. Fibrous Dysplasia – an Update. Journal of Bone and Soft Tissue Tumors Jan-Apr 2016;2(1): 39-43. |

Like this:
Like Loading...